RSVinA549
scottzijiezhang
2018-08-08
Last updated: 2019-05-06
workflowr checks: (Click a bullet for more information)-
✔ R Markdown file: up-to-date
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
-
✔ Environment: empty
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
-
✔ Seed:
set.seed(20190123)The command
set.seed(20190123)was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible. -
✔ Session information: recorded
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
-
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.✔ Repository version: 295e8d4
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can usewflow_publishorwflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.Untracked files: Untracked: analysis/RSVmutant.Rmd Untracked: data/A549_jointPeak_readCount.txt Untracked: data/Hela_jointPeak_readCount.txt Untracked: docs/figure/RSVmutant.Rmd/ Unstaged changes: Modified: analysis/_site.yml
Expand here to see past versions:
vgRNA
library(m6Amonster)
vgRNA <-c("RSVvgRNA1","RSVvgRNA2")
gtf <- "~/Database/genome/RSV/GFP.RSV_gene.gtf"
RSV.A549 <- countReads(samplenames = paste(vgRNA,"align_RSV",sep = "." ),
gtf = gtf,
bamFolder = "/home/zijiezhang/RSV/201803/bam_files",
outputDir = "/home/zijiezhang/RSV/201803",
modification = "m6A",
threads = 2,saveOutput =F,
binSize = 30
)Reading gtf file to obtain gene model
Filter out ambiguous model...
Gene model obtained from gtf file...
counting reads for each genes, this step may takes a few hours....
Hyper-thread registered: TRUE
Using 2 thread(s) to count reads in continuous bins...
Time used to count reads: 0.108239122231801 mins... Report the peaks on vgRNA.
RSV.A549 <- m6Amonster:::callPeakBinomial(RSV.A549,min_counts = 10, threads = 10)
vgRNA_peak <- reportConsistentPeak(readsOut = RSV.A549,samplenames = paste(vgRNA,"align_RSV",sep = "." ))Reporting peak concsistent in all samples for
RSVvgRNA1.align_RSV RSVvgRNA2.align_RSV
Hyper-thread registered: TRUE
Using 1 thread(s) to report merged report...
Time used to report peaks: 0.0257289528846741 mins... annotation <- read.table("~/Database/genome/RSV/GFP.RSV_annotation.txt",sep = "\t",header = T)
anno.gr <- makeGRangesFromDataFrame(annotation,keep.extra.columns = T)
vgRNA_gr <- makeGRangesFromDataFrame(vgRNA_peak)
anno.vgRNA <- as.data.frame(findOverlaps(vgRNA_gr, anno.gr, ignore.strand = T) )
vgRNA_peak$name <- as.character(vgRNA_peak$name)
vgRNA_peak$name [anno.vgRNA$queryHits] <- as.character(annotation[anno.vgRNA$subjectHits,"gene"])
write.table(dplyr::filter(vgRNA_peak,score<1e-20),file = "~/RSV/RSV_m6Aseq_analysis/data/RSVvgRNA_A549_peaks.xls", sep = "\t",col.names = T,row.names = F,quote = F)Plot the coverage of vgRNA.
library(MyTools)
RSV.A549_plot <- gtfToGeneModel( "~/Database/genome/RSV/GFP.RSV.gtf")
plotVirusCov(RSV.A549$bamPath.ip, RSV.A549$bamPath.input ,RSV.A549_plot,libraryType = "opposite",center = mean,annotation)+scale_fill_discrete(name = "IP",labels = c("Genome","anti-Genome"))+ xlab("Genome location") + ylab("Normalized coverage") + scale_colour_discrete(name = "INPUT",labels = c("Genome","anti-Genome"))+theme(legend.text = element_text(face = "bold",size = 18), legend.title = element_text(face = "bold",size = 20),axis.text = element_text(face = "bold",size = 18),axis.title = element_text(face = "bold",size = 20) )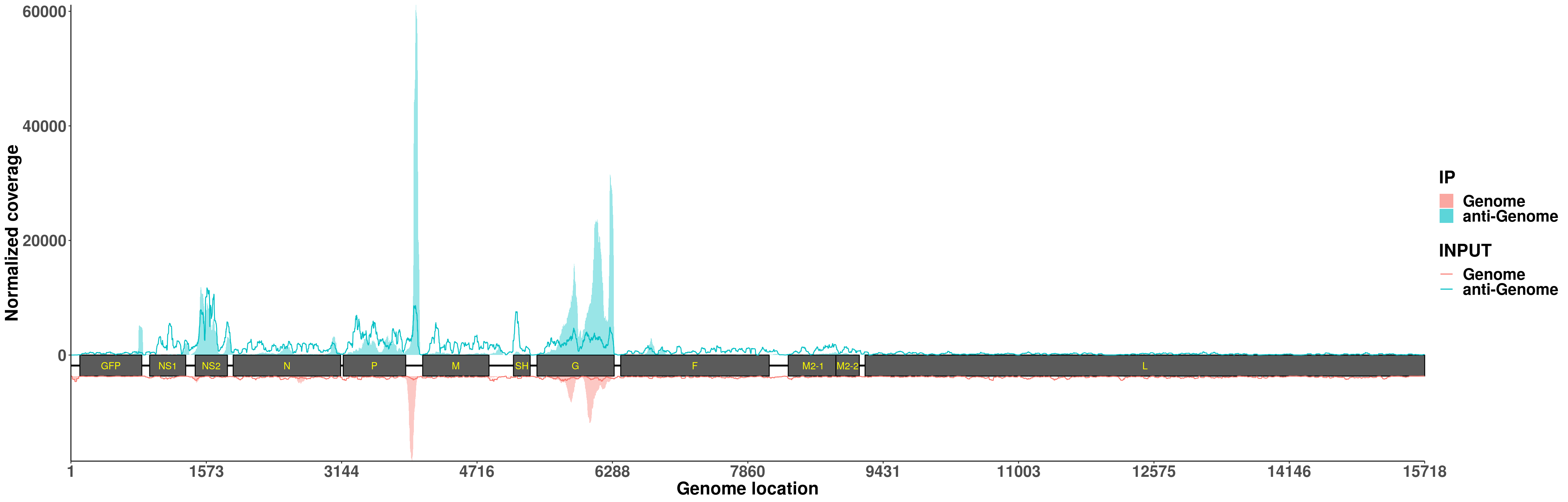
Expand here to see past versions of unnamed-chunk-3-1.png:
| Version | Author | Date |
|---|---|---|
| 295e8d4 | scottzijiezhang | 2019-01-23 |
Infected sample
infected <- c("RSVinfect1","RSVinfect2","mutRSVinfect1","mutRSVinfect2")
RSV_infect <- countReads(samplenames = paste(infected,"align_RSV",sep = "." ),
gtf = gtf,
bamFolder = "/home/zijiezhang/RSV/201803/bam_files",
outputDir = "/home/zijiezhang/RSV/201803",
modification = "m6A",
threads = 2,saveOutput = F,
binSize = 30
)Reading gtf file to obtain gene model
Filter out ambiguous model...
Gene model obtained from gtf file...
counting reads for each genes, this step may takes a few hours....
Hyper-thread registered: TRUE
Using 2 thread(s) to count reads in continuous bins...
Time used to count reads: 0.719945641358693 mins... RSV_infect <- m6Amonster:::callPeakBinomial(RSV_infect,threads = 10)Report peaks for infected samples
WT_peak <- reportConsistentPeak(RSV_infect,samplenames = paste(infected,"align_RSV",sep = "." )[1:2])Reporting peak concsistent in all samples for
RSVinfect1.align_RSV RSVinfect2.align_RSV
Hyper-thread registered: TRUE
Using 1 thread(s) to report merged report...
Time used to report peaks: 0.00843370358149211 mins... ## annotate peak
WT_peak_gr <- makeGRangesFromDataFrame(WT_peak)
anno.WT <- as.data.frame(findOverlaps(WT_peak_gr, anno.gr, ignore.strand = T,minoverlap = 100) )
WT_peak$name <- as.character(WT_peak$name)
WT_peak$name [anno.WT$queryHits] <- as.character(annotation[anno.WT$subjectHits,"gene"])
write.table(dplyr::filter(WT_peak,score<1e-5),file = "~/RSV/RSV_m6Aseq_analysis/data/RSVinfected_A549_peaks.xls", sep = "\t",col.names = T,row.names = F,quote = F)Plot WT coverage
plotVirusCov(RSV_infect$bamPath.ip[1:2],RSV_infect$bamPath.input[1:2] ,RSV.A549_plot,libraryType = "opposite",center = mean,annotation,hideStrand = "-")+scale_fill_discrete(name = "IP",labels = c("anti-Genome/mRNA")) + xlab("Genome location") + ylab("Normalized coverage")+ scale_colour_discrete(name = "INPUT",labels = c("anti-Genome/mRNA"))+theme(legend.text = element_text(face = "bold",size = 18), legend.title = element_text(face = "bold",size = 20),axis.text = element_text(face = "bold",size = 18),axis.title = element_text(face = "bold",size = 20) )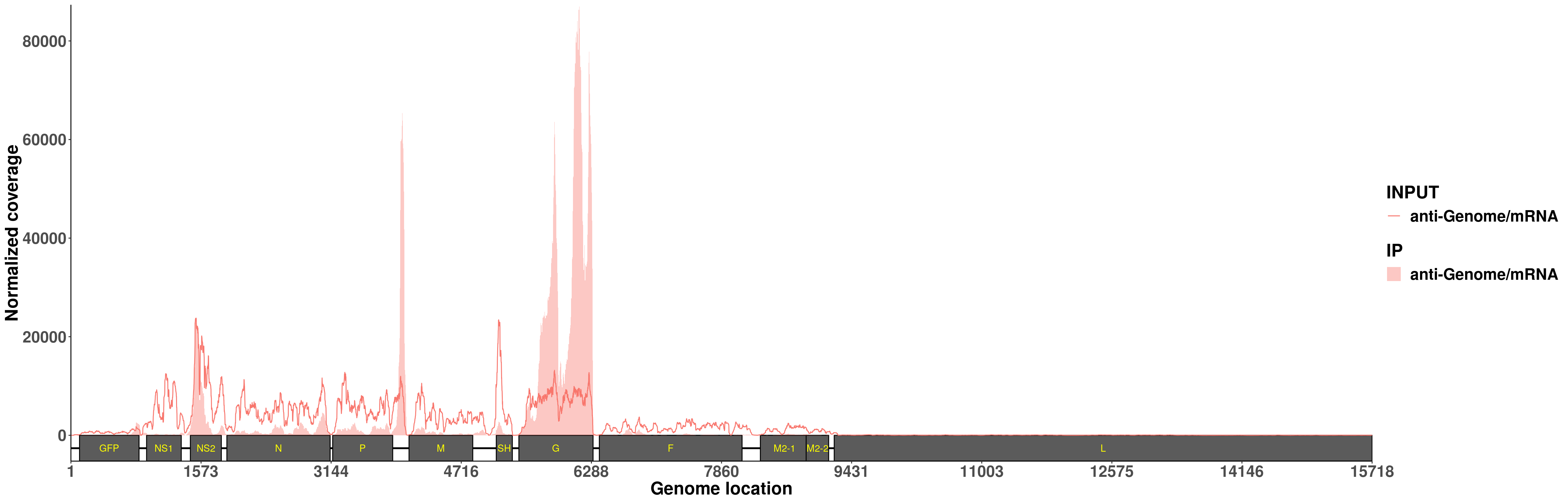
Expand here to see past versions of unnamed-chunk-6-1.png:
| Version | Author | Date |
|---|---|---|
| 295e8d4 | scottzijiezhang | 2019-01-23 |
Session information
sessionInfo()R version 3.5.3 (2019-03-11)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 17.10
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/openblas/libblas.so.3
LAPACK: /usr/lib/x86_64-linux-gnu/libopenblasp-r0.2.20.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] grid stats4 parallel stats graphics grDevices utils
[8] datasets methods base
other attached packages:
[1] MyTools_0.0.0 ChIPseeker_1.18.0
[3] Guitar_1.20.0 bindrcpp_0.2.2
[5] m6Amonster_0.1.5 RcppArmadillo_0.9.200.5.0
[7] Rcpp_1.0.0 reshape2_1.4.3
[9] GenomicAlignments_1.18.0 SummarizedExperiment_1.12.0
[11] DelayedArray_0.8.0 BiocParallel_1.16.1
[13] matrixStats_0.54.0 rtracklayer_1.42.1
[15] doParallel_1.0.14 iterators_1.0.10
[17] foreach_1.4.4 ggplot2_3.1.0
[19] Rsamtools_1.34.0 Biostrings_2.50.1
[21] XVector_0.22.0 GenomicFeatures_1.34.1
[23] AnnotationDbi_1.44.0 Biobase_2.42.0
[25] GenomicRanges_1.34.0 GenomeInfoDb_1.18.1
[27] IRanges_2.16.0 S4Vectors_0.20.1
[29] BiocGenerics_0.28.0
loaded via a namespace (and not attached):
[1] backports_1.1.2
[2] fastmatch_1.1-0
[3] workflowr_1.1.1
[4] plyr_1.8.4
[5] igraph_1.2.2
[6] lazyeval_0.2.1
[7] splines_3.5.3
[8] gridBase_0.4-7
[9] urltools_1.7.1
[10] digest_0.6.18
[11] htmltools_0.3.6
[12] GOSemSim_2.8.0
[13] viridis_0.5.1
[14] GO.db_3.7.0
[15] gdata_2.18.0
[16] magrittr_1.5
[17] memoise_1.1.0
[18] cluster_2.0.7-1
[19] R.utils_2.7.0
[20] enrichplot_1.2.0
[21] prettyunits_1.0.2
[22] colorspace_1.4-0
[23] blob_1.1.1
[24] ggrepel_0.8.0
[25] dplyr_0.7.8
[26] crayon_1.3.4
[27] RCurl_1.95-4.11
[28] jsonlite_1.5
[29] TxDb.Hsapiens.UCSC.hg19.knownGene_3.2.2
[30] bindr_0.1.1
[31] ape_5.2
[32] glue_1.3.0
[33] gtable_0.2.0
[34] zlibbioc_1.28.0
[35] UpSetR_1.3.3
[36] scales_1.0.0
[37] DOSE_3.8.0
[38] DBI_1.0.0
[39] plotrix_3.7-4
[40] viridisLite_0.3.0
[41] progress_1.2.0
[42] units_0.6-1
[43] gridGraphics_0.3-0
[44] bit_1.1-14
[45] europepmc_0.3
[46] httr_1.3.1
[47] fgsea_1.8.0
[48] gplots_3.0.1
[49] RColorBrewer_1.1-2
[50] pkgconfig_2.0.2
[51] XML_3.98-1.16
[52] R.methodsS3_1.7.1
[53] farver_1.1.0
[54] ggplotify_0.0.3
[55] tidyselect_0.2.5
[56] labeling_0.3
[57] rlang_0.3.1
[58] munsell_0.5.0
[59] tools_3.5.3
[60] RSQLite_2.1.1
[61] ggridges_0.5.1
[62] evaluate_0.12
[63] stringr_1.3.1
[64] yaml_2.2.0
[65] knitr_1.20
[66] bit64_0.9-7
[67] caTools_1.17.1.1
[68] purrr_0.2.5
[69] ggraph_1.0.2
[70] nlme_3.1-137
[71] whisker_0.3-2
[72] R.oo_1.22.0
[73] DO.db_2.9
[74] xml2_1.2.0
[75] biomaRt_2.38.0
[76] compiler_3.5.3
[77] tibble_2.0.1
[78] tweenr_1.0.0
[79] stringi_1.2.4
[80] lattice_0.20-38
[81] Matrix_1.2-15
[82] vegan_2.5-3
[83] permute_0.9-4
[84] pillar_1.3.1
[85] triebeard_0.3.0
[86] data.table_1.11.8
[87] cowplot_0.9.3
[88] bitops_1.0-6
[89] qvalue_2.14.0
[90] R6_2.3.0
[91] vcfR_1.8.0
[92] KernSmooth_2.23-15
[93] gridExtra_2.3
[94] codetools_0.2-16
[95] boot_1.3-20
[96] MASS_7.3-51.1
[97] gtools_3.8.1
[98] assertthat_0.2.0
[99] rprojroot_1.3-2
[100] withr_2.1.2
[101] pinfsc50_1.1.0
[102] GenomeInfoDbData_1.2.0
[103] mgcv_1.8-26
[104] hms_0.4.2
[105] rmarkdown_1.10
[106] rvcheck_0.1.1
[107] git2r_0.23.0
[108] ggforce_0.1.3 This reproducible R Markdown analysis was created with workflowr 1.1.1