T2D
Zijie Zhang
2018-08-21
Count reads
library(RADAR)
samplenames = c("Ctl1","Ctl4","Ctl8","Ctl16","Ctl17","Ctl18","Ctl20","T2D1","T2D4","T2D5","T2D6","T2D7","T2D9","T2D11","T2D12")
RADAR <- countReads(samplenames = samplenames,
gtf = "~/Database/genome/hg38/hg38_UCSC.gtf",
bamFolder = "~/Rohit_T2D/bam_files",
outputDir = "~/Rohit_T2D",
modification = "IP",
threads = 25
)
monster <- RADARSummary of read count
library(RADAR)
library(qvalue)
#load("~/Rohit_T2D/RADAR.RData")
load("~/Tools/RADARmannual/data/T2D_MeRIP.RADAR.RData")
summary(RADAR)## MeRIP.RADAR dataset of 15 samples.
## Read count quantified in 50-bp consecutive bins on the transcript.
## The total read count for Input and IP samples are (Million reads):
## Ctl1 Ctl4 Ctl8 Ctl16 Ctl17 Ctl18 Ctl20 T2D1 T2D4 T2D5 T2D6
## Input 18.32 17.75 17.92 11.51 15.12 25.92 20.30 16.80 16.11 14.15 17.20
## IP 30.29 25.11 21.91 23.20 21.41 31.65 24.75 27.73 26.63 28.46 22.54
## T2D7 T2D9 T2D11 T2D12
## Input 14.34 17.76 19.78 18.78
## IP 36.10 27.43 22.05 19.40
## Input gene level read count available.
## There are 4 predictor variables/covariates. Can access by function variable(MeRIPdata).RADAR <- normalizeLibrary(RADAR)
RADAR <- adjustExprLevel(RADAR )
RADAR <- filterBins(RADAR, minCountsCutOff = 15)
RADAR_pos <- RADAR::adjustExprLevel(RADAR, adjustBy = "peak")
RADAR_pos <- filterBins(RADAR_pos, minCountsCutOff = 15)
library(rafalib)
X2 <- as.fumeric(c("A","A","A","B","B","A","A","A","A","B","A","B","B","A","A"))-1 # batch as covariates
X3 <- as.fumeric(c("A","A","A","A","A","C","C","A","A","A","A","A","A","A","C"))-1 # batch as covariates
X4 <- as.fumeric(c("M","F","M","M","M","F","M","M","M","F","M","M","M","M","F"))-1 # sex as covariates
variable(RADAR)<- data.frame( disease = c(rep("Ctl",7),rep("T2D",8)), X2 = X2, X3 = X3, X4 = X4 )INPUT read count
Local INPUT read count
Plot distribution of number of reads in each 50 bp bins
hist(log10(rowMeans(RADAR@reads[rowMeans(RADAR@reads[,grep("input",colnames(RADAR@reads))])>1,grep("input",colnames(RADAR@reads))]) ),xlab = "log10 read count",main = "Distribution of INPUT read count in bins",xlim = c(0,3), col =rgb(0.2,0.2,0.2,0.5),cex.main = 2,cex.axis =2,cex.lab=2)
axis(side = 1, lwd = 2,cex.axis =2)
axis(side = 2, lwd = 2,cex.axis =2)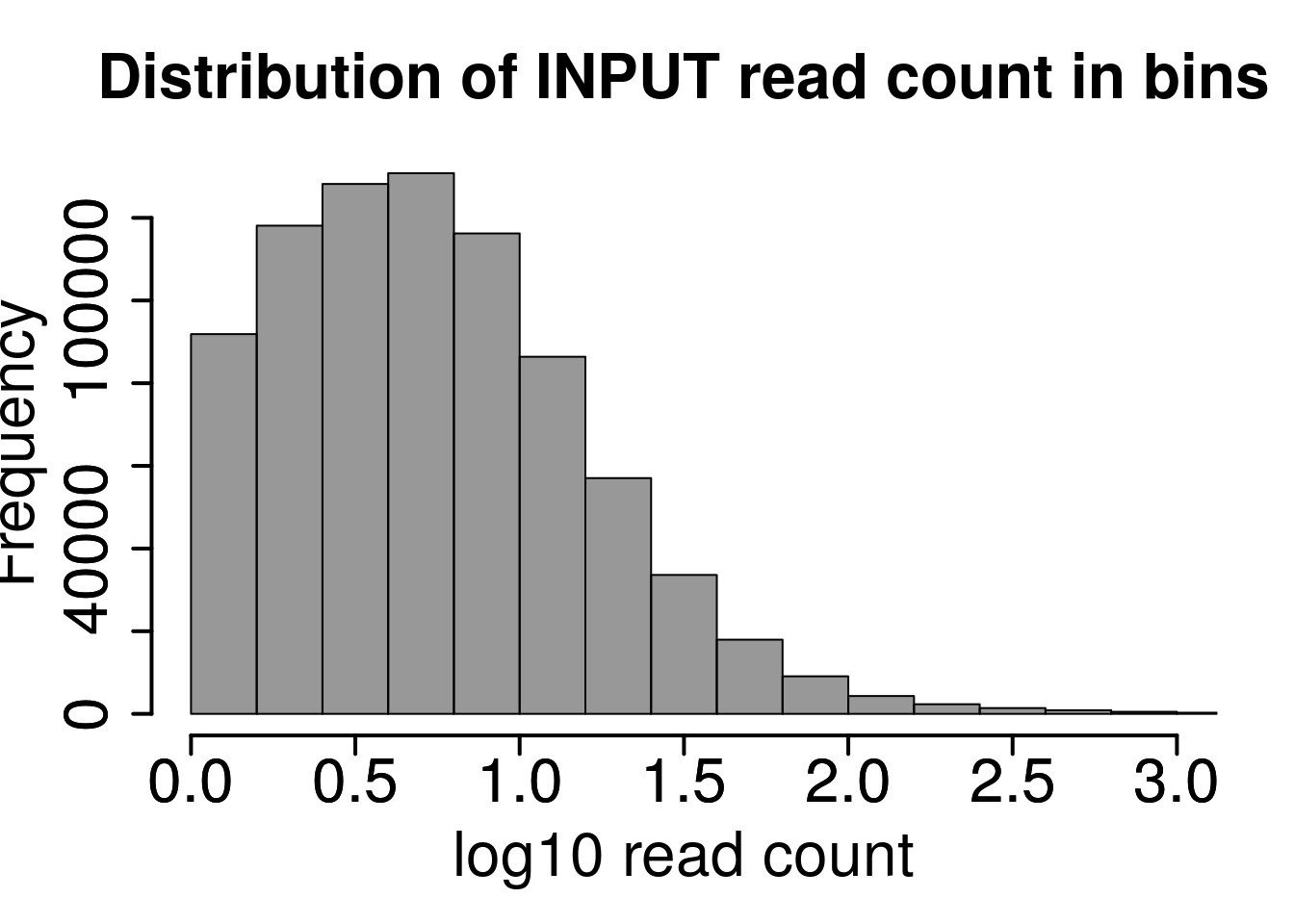
local read count V.S. geneSum
Compute the within group variability of INPUT geneSum VS INPUT local read count.
vvar.coef <- function(x){sd(as.numeric(x))/mean(as.numeric(x))}
## filter expressed genes
geneSum <- RADAR@geneSum[rowSums(RADAR@geneSum)>16,]
## within group variability
relative.var <- t( apply(geneSum,1,tapply,unlist(variable(RADAR)[,1]),var.coef) )
geneSum.var <- c(relative.var)
## For each gene used above, random sample a 50bp bin within this gene
set.seed(1)
r.bin50 <- tapply(rownames(RADAR@norm.input)[which(RADAR@geneBins$gene %in% rownames(geneSum))],as.character( RADAR@geneBins$gene[which(RADAR@geneBins$gene %in% rownames(geneSum))]) ,function(x){
n <- sample(1:length(x),1)
return(x[n])
})
relative.var <- apply(RADAR@norm.input[r.bin50,],1,tapply,unlist(variable(RADAR)[,1]),var.coef)
bin50.var <- c(relative.var)
bin50.var <- bin50.var[!is.na(bin50.var)]
## 100bp bins
r.bin100 <- tapply(rownames(RADAR@norm.input)[which(RADAR@geneBins$gene %in% rownames(geneSum))],as.character( RADAR@geneBins$gene[which(RADAR@geneBins$gene %in% rownames(geneSum))]) ,function(x){
n <- sample(1:(length(x)-2),1)
return(x[n:(n+1)])
})
relative.var <- lapply(r.bin100, function(x){ tapply( colSums(RADAR@norm.input[x,]),unlist(variable(RADAR)[,1]),var.coef ) })
bin100.var <- unlist(relative.var)
bin100.var <- bin100.var[!is.na(bin100.var)]
## 200bp bins
r.bin200 <- tapply(rownames(RADAR@norm.input)[which(RADAR@geneBins$gene %in% rownames(geneSum))],as.character( RADAR@geneBins$gene[which(RADAR@geneBins$gene %in% rownames(geneSum))]) ,function(x){
if(length(x)>4){
n <- sample(1:(length(x)-3),1)
return(x[n:(n+3)])
}else{
return(NULL)
}
})
r.bin200 <- r.bin200[which(!unlist(lapply(r.bin200,is.null)) ) ]
relative.var <- lapply(r.bin200, function(x){ tapply( colSums(RADAR@norm.input[x,]),unlist(variable(RADAR)[,1]),var.coef ) })
bin200.var <- unlist(relative.var)
bin200.var <- bin200.var[!is.na(bin200.var)]
relative.var <- data.frame(group=c(rep("geneSum",length(geneSum.var)),rep("bin-50bp",length(bin50.var)),rep("bin-100bp",length(bin100.var)),rep("bin-200bp",length(bin200.var))), variance=c(geneSum.var,bin50.var,bin100.var,bin200.var))ggplot(relative.var,aes(variance,colour=group))+geom_density()+xlab("Relative variation")+
theme_bw() + theme(panel.border = element_blank(), panel.grid.major = element_blank(),
panel.grid.minor = element_blank(), axis.line = element_line(colour = "black",size = 1),
axis.title.x=element_text(size=20, face="bold", hjust=0.5,family = "arial"),
axis.title.y=element_text(size=20, face="bold", vjust=0.4, angle=90,family = "arial"),
legend.position = c(0.88,0.88),legend.title=element_blank(),legend.text = element_text(size = 14, face = "bold",family = "arial"),
axis.text = element_text(size = 15,face = "bold",family = "arial",colour = "black") )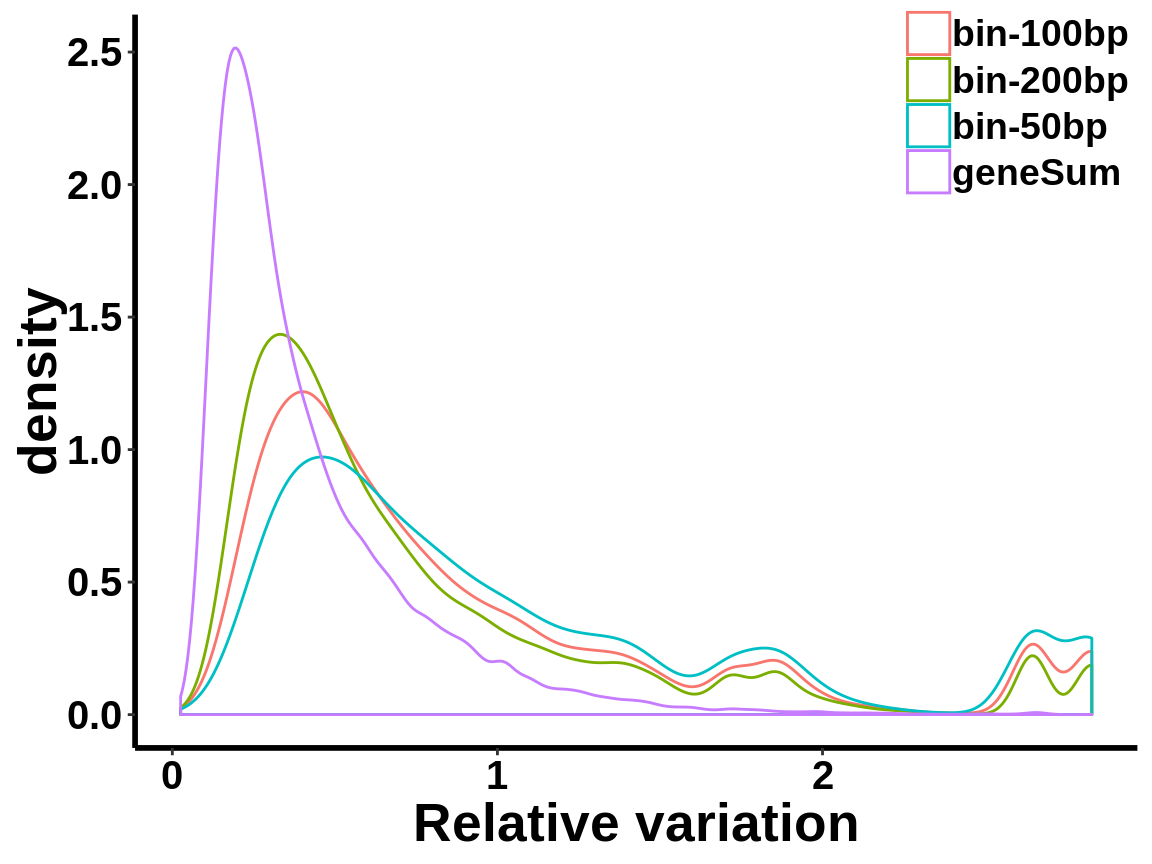
Compare MeRIP-seq IP data with regular RNA-seq data
var.coef <- function(x){sd(as.numeric(x))/mean(as.numeric(x))}
ip_coef_var <- t(apply(RADAR@ip_adjExpr_filtered,1,tapply,unlist(variable(RADAR)[,1]),var.coef))
ip_coef_var <- ip_coef_var[!apply(ip_coef_var,1,function(x){return(any(is.na(x)))}),]
#hist(c(ip_coef_var),main = "M14KO mouse liver\n m6A-IP",xlab = "within group coefficient of variation",breaks = 50)
###
gene_coef_var <- t(apply(RADAR@geneSum,1,tapply,unlist(variable(RADAR)[,1]),var.coef))
gene_coef_var <- gene_coef_var[!apply(gene_coef_var,1,function(x){return(any(is.na(x)))}),]
#hist(c(gene_coef_var),main = "M14KO mouse liver\n RNA-seq",xlab = "within group coefficient of variation",breaks = 50)
coef_var <- list('RNA-seq'=c(gene_coef_var),'m6A-IP'=c(ip_coef_var))
nn<-sapply(coef_var, length)
rs<-cumsum(nn)
re<-rs-nn+1
grp <- factor(rep(names(coef_var), nn), levels=names(coef_var))
coef_var.df <- data.frame(coefficient_var = c(c(gene_coef_var),c(ip_coef_var)),label = grp)ggplot(data = coef_var.df,aes(coefficient_var,colour = grp))+geom_density()+ theme_bw() + theme(panel.border = element_blank(), panel.grid.major = element_blank(),
panel.grid.minor = element_blank(), axis.line = element_line(colour = "black",size = 1),
axis.title.x=element_text(size=20, face="bold", hjust=0.5,family = "arial"),
axis.title.y=element_text(size=20, face="bold", vjust=0.4, angle=90,family = "arial"),
legend.position = c(0.9,0.9),legend.title=element_blank(),legend.text = element_text(size = 18, face = "bold",family = "arial"),
axis.text = element_text(size = 15,face = "bold",family = "arial",colour = "black") )+
xlab("Coefficient of variation")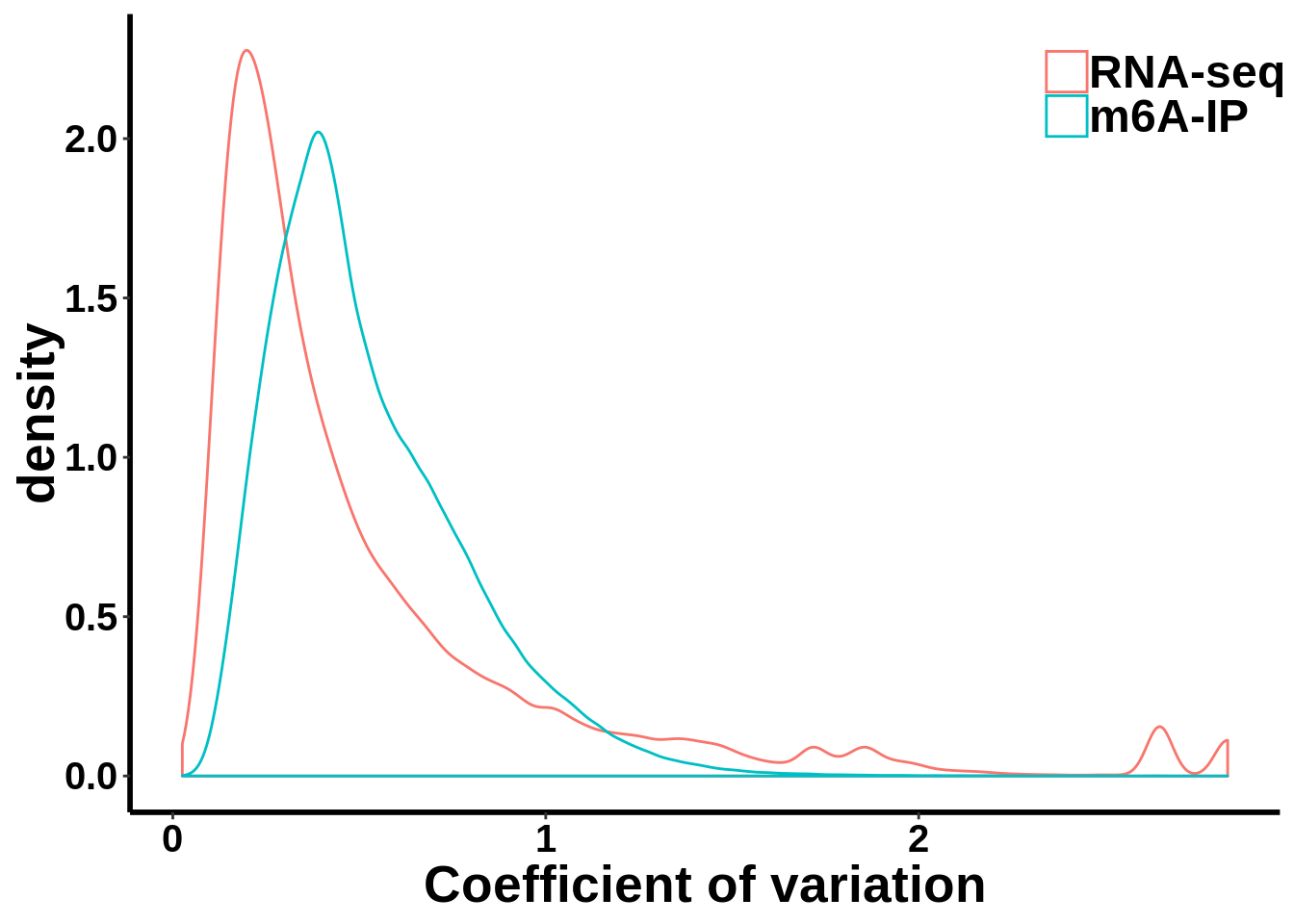
Check mean variance relationship of the data.
ip_var <- t(apply(RADAR@ip_adjExpr_filtered,1,tapply,unlist(variable(RADAR)[,1]),var))
#ip_var <- ip_coef_var[!apply(ip_var,1,function(x){return(any(is.na(x)))}),]
ip_mean <- t(apply(RADAR@ip_adjExpr_filtered,1,tapply,unlist(variable(RADAR)[,1]),mean))
###
gene_var <- t(apply(RADAR@geneSum,1,tapply,unlist(variable(RADAR)[,1]),var))
#gene_var <- gene_var[!apply(gene_var,1,function(x){return(any(is.na(x)))}),]
gene_mean <- t(apply(RADAR@geneSum,1,tapply,unlist(variable(RADAR)[,1]),mean))
all_var <- list('RNA-seq'=c(gene_var),'m6A-IP'=c(ip_var))
nn<-sapply(all_var, length)
rs<-cumsum(nn)
re<-rs-nn+1
group <- factor(rep(names(all_var), nn), levels=names(all_var))
all_var.df <- data.frame(variance = c(c(gene_var),c(ip_var)),mean= c(c(gene_mean),c(ip_mean)),label = group)ggplot(data = all_var.df,aes(x=mean,y=variance,colour = group,shape = group))+geom_point(size = I(0.2))+stat_smooth(se = T,show.legend = F)+stat_smooth(se = F)+theme_bw() +xlab("Mean")+ylab("Variance") +theme(panel.border = element_blank(), panel.grid.major = element_blank(),
panel.grid.minor = element_blank(), axis.line = element_line(colour = "black",size = 1),
axis.title.x=element_text(size=22, face="bold", hjust=0.5,family = "arial"),
axis.title.y=element_text(size=22, face="bold", vjust=0.4, angle=90,family = "arial"),
legend.position = c(0.85,0.95),legend.title=element_blank(),legend.text = element_text(size = 20, face = "bold",family = "arial"),
axis.text = element_text(size = 15,face = "bold",family = "arial",colour = "black") ) +
scale_x_continuous(limits = c(0,10000))+scale_y_continuous(limits = c(0,3e7))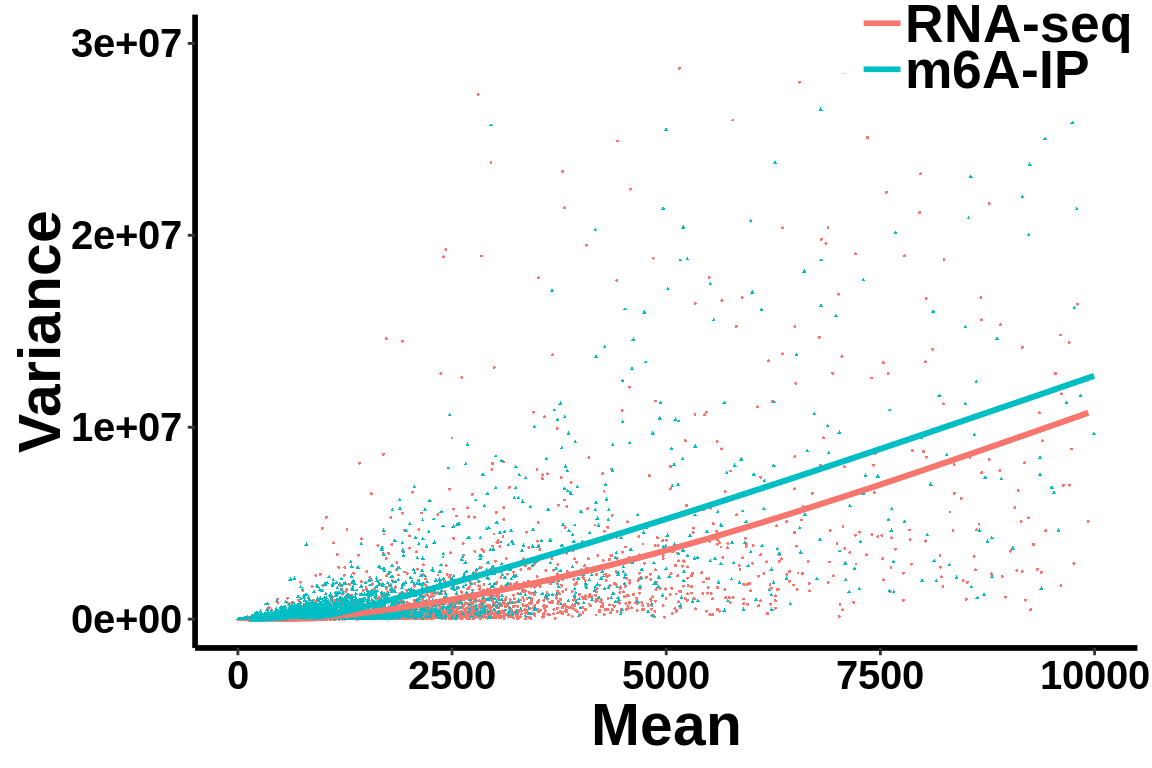
inputCount <- as.data.frame(RADAR@reads[,1:length(RADAR@samplenames)])
logInputCount <- as.data.frame(log( RADAR@geneSum ) )
ggplot(logInputCount, aes(x = `Ctl18` , y = `T2D6` ))+geom_point()+geom_abline(intercept = 0,slope = 1, lty = 2, colour = "red")+xlab(paste("Log counts Ctl18") )+ylab(paste("Log counts T2D6") )+theme_classic(base_line_size = 0.8)+theme(axis.title = element_text(size = 12,face = "bold"), axis.text = element_text(size = 10,face = "bold",colour = "black") )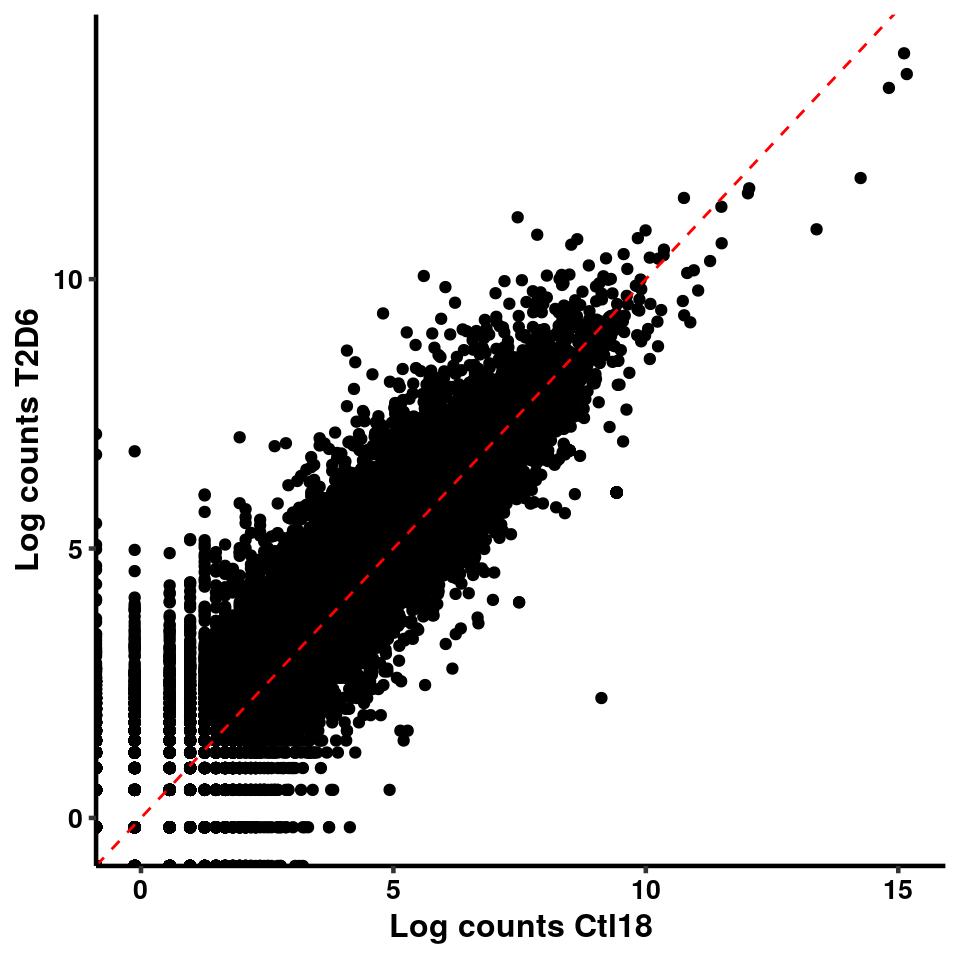
ggplot(logInputCount, aes(x = `Ctl18` , y = `T2D5` ))+geom_point()+geom_abline(intercept = 0,slope = 1, lty = 2, colour = "red")+xlab(paste("Log counts Ctl18") )+ylab(paste("Log counts T2D5") )+theme_classic(base_line_size = 0.8)+theme(axis.title = element_text(size = 12,face = "bold"), axis.text = element_text(size = 10,face = "bold",colour = "black") )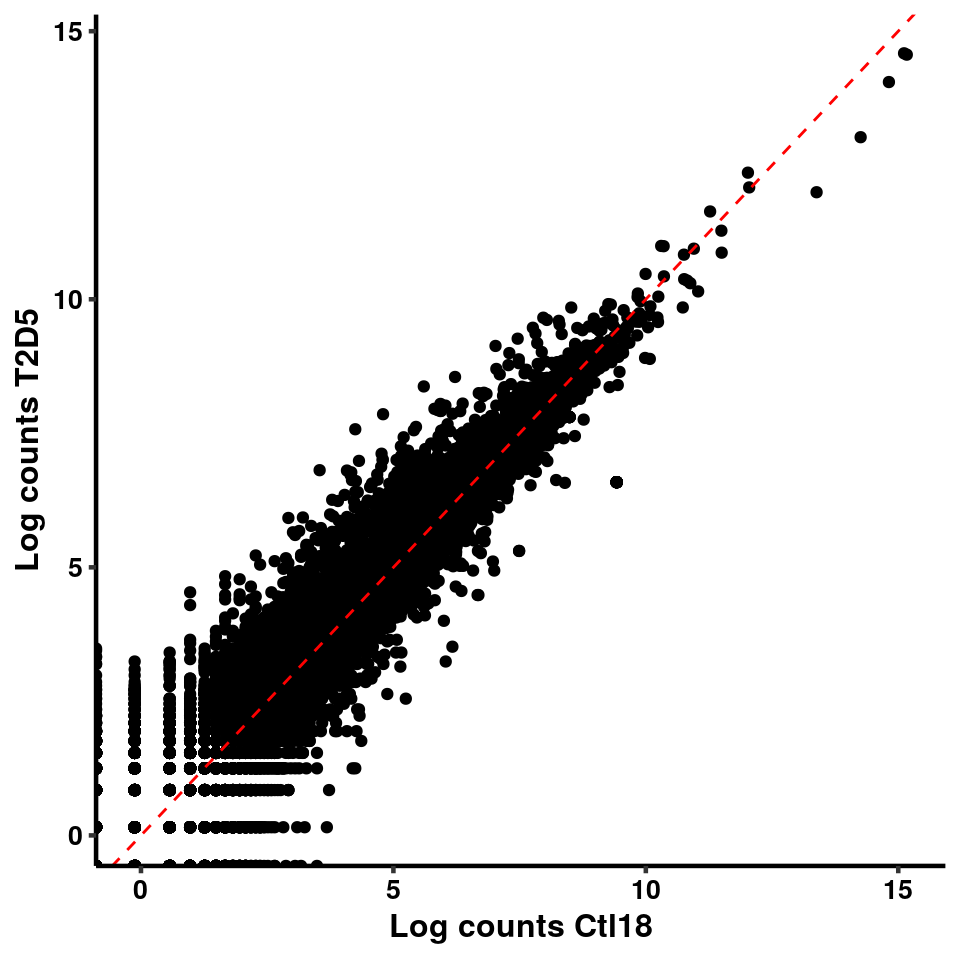
PCA analysis
After geting the filtered and normalized IP read count for testing, we can use PCA analysis to check unkown variation.
top_bins <- RADAR@ip_adjExpr_filtered[order(rowMeans(RADAR@ip_adjExpr_filtered))[1:15000],]We plot PCA colored by Ctl vs T2D
plotPCAfromMatrix(top_bins,group = unlist(variable(RADAR)[,1]))+scale_color_discrete(name = "Group")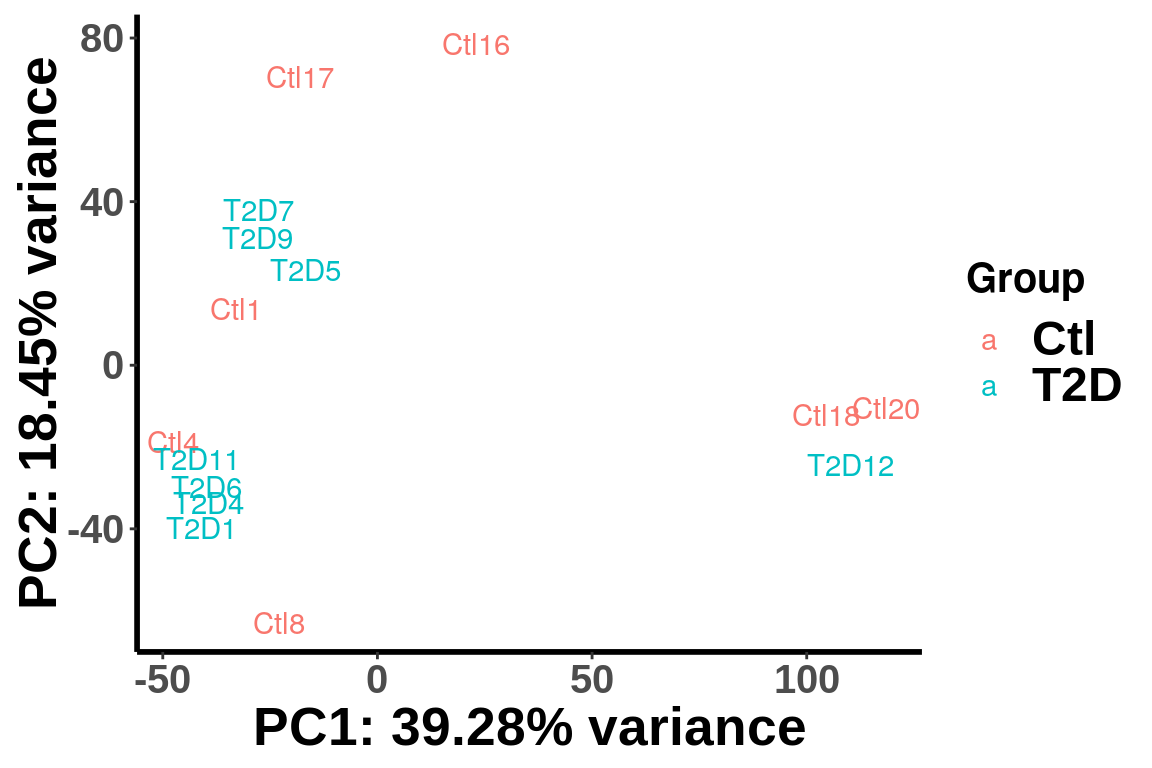
(Norm by position)
plotPCAfromMatrix(RADAR_pos@ip_adjExpr_filtered[order(rowMeans(RADAR_pos@ip_adjExpr_filtered))[1:15000],],group = unlist(variable(RADAR)[,1]) )+scale_color_discrete(name = "Group")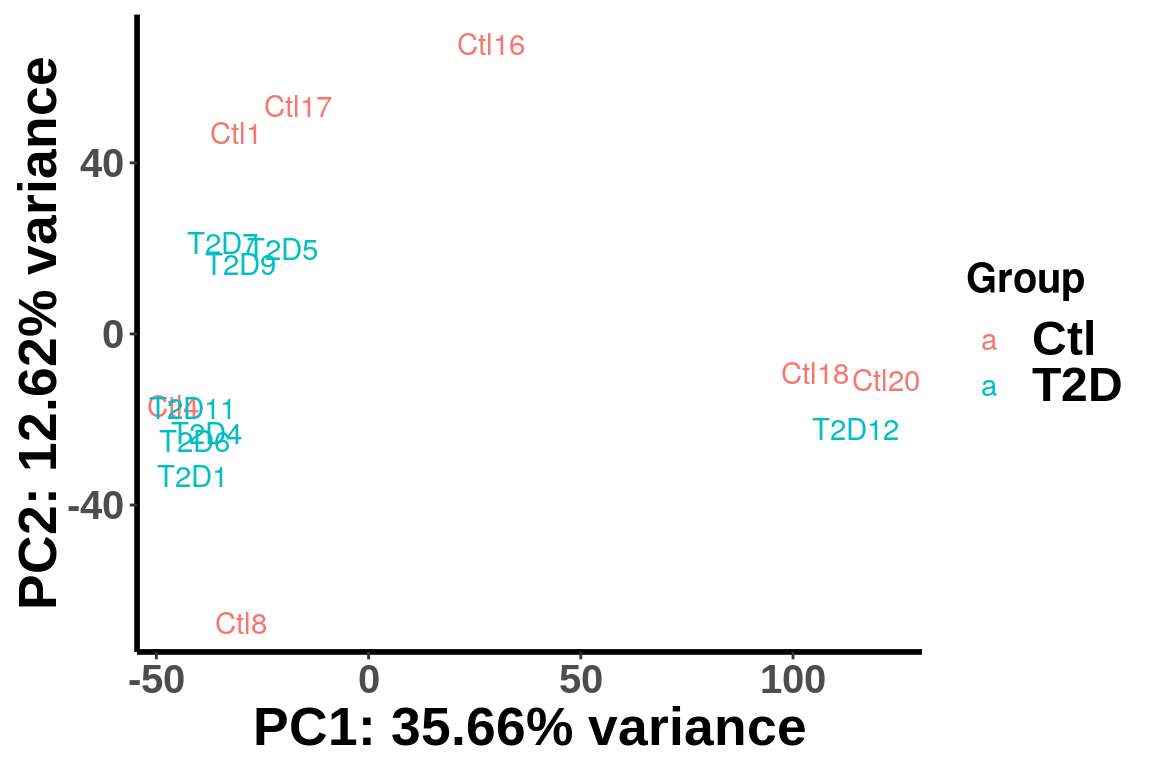
To check if samples are separated by different sequencing lanes, label the samples by lane batch.
batch <- c("A","A","A","B","B","C","C","A","A","B","A","B","B","A","C")
plotPCAfromMatrix(top_bins,group = paste0(batch))+scale_color_discrete(name = "Batch")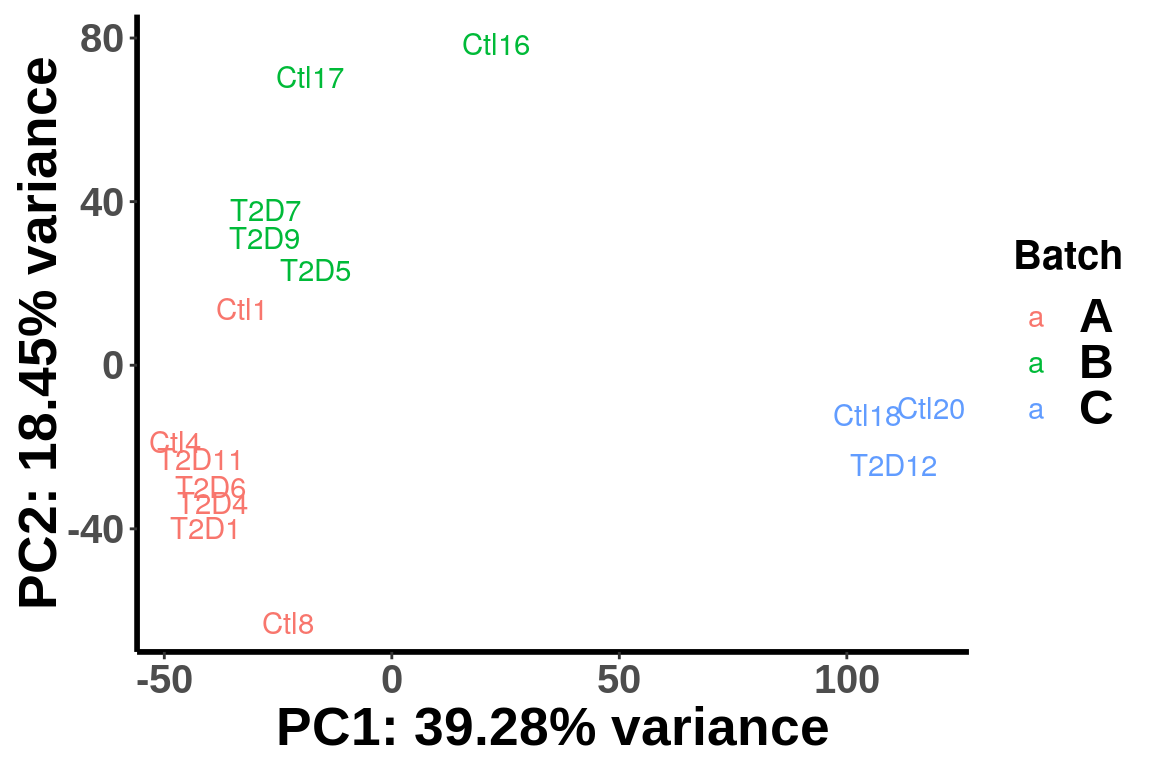
We can see that the samples are clearly separated by batches and we need to account for batch effect in the test. Thus we incorporate batch and gender as known covariates in the test.
Check the PCA after r egressing out the batch and gender.
library(rafalib)
X2 <- as.fumeric(c("A","A","A","B","B","A","A","A","A","B","A","B","B","A","A"))-1 # batch as covariates
X3 <- as.fumeric(c("A","A","A","A","A","C","C","A","A","A","A","A","A","A","C"))-1 # batch as covariates
X4 <- as.fumeric(c("M","F","M","M","M","F","M","M","M","F","M","M","M","M","F"))-1 # sex as covariates
registerDoParallel(cores = 10)
m6A.cov.out <- foreach(i = 1:nrow(top_bins), .combine = rbind) %dopar% {
Y = top_bins[i,]
resi <- residuals( lm(log(Y+1) ~ X2 + X3 + X4 ) )
resi
}
rm(list=ls(name=foreach:::.foreachGlobals), pos=foreach:::.foreachGlobals)
rownames(m6A.cov.out) <- rownames(top_bins)plotPCAfromMatrix(m6A.cov.out,unlist(variable(RADAR)[,1]),loglink = FALSE)+scale_color_discrete(name = "Group")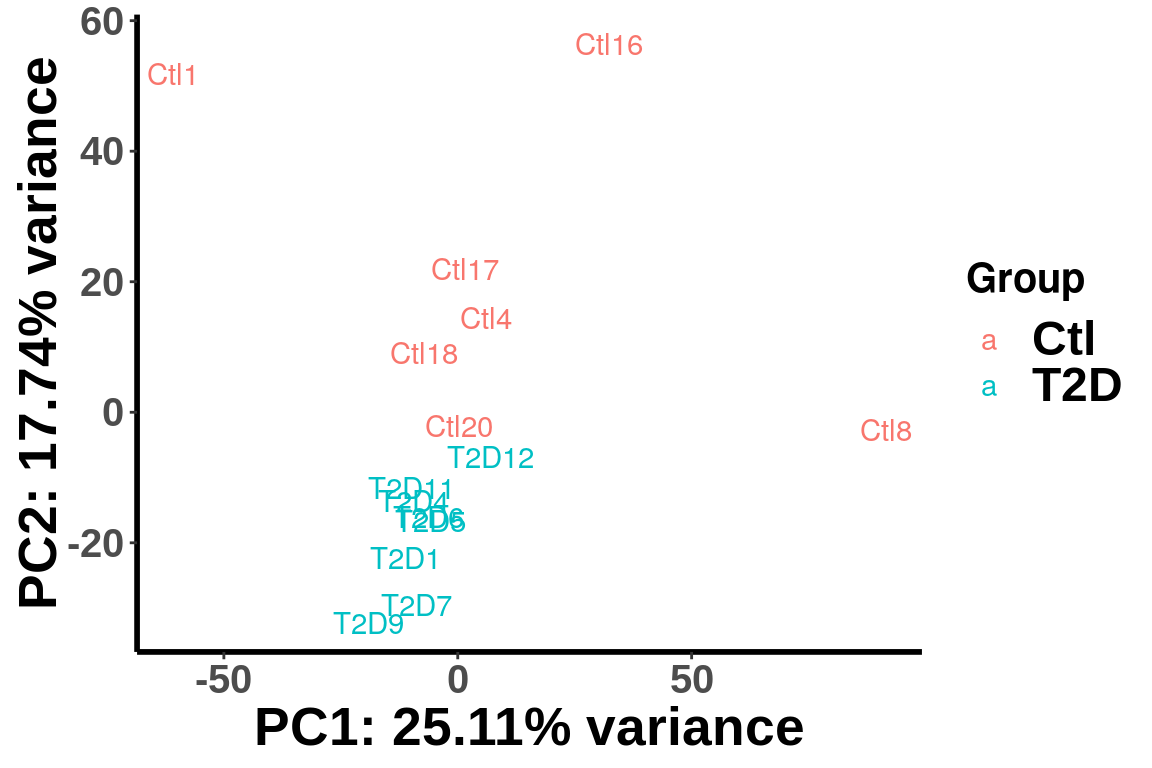
After regressing out the covariates, the samples are separated by diseases status in the PCA plot.
Run PoissonGamma test
cov <- cbind(X2,X3,X4)
RADAR <- diffIP_parallel(RADAR, thread = 20 )
RADAR_pos <- diffIP_parallel(RADAR_pos,thread = 25 )Other method
## fisher's exact test
fisherTest <- function( control_ip, treated_ip, control_input, treated_input, thread ){
registerDoParallel(cores = thread)
testResult <- foreach(i = 1:nrow(control_ip) , .combine = rbind )%dopar%{
tmpTest <- fisher.test( cbind( c( rowSums(treated_ip)[i], rowSums(treated_input)[i] ), c( rowSums(control_ip)[i], rowSums(control_input)[i] ) ), alternative = "two.sided" )
data.frame( logFC = log(tmpTest$estimate), pvalue = tmpTest$p.value )
}
rm(list=ls(name=foreach:::.foreachGlobals), pos=foreach:::.foreachGlobals)
testResult$fdr <- p.adjust(testResult$pvalue, method = "fdr")
return(testResult)
}
## wrapper for the MetDiff test
MeTDiffTest <- function( control_ip, treated_ip, control_input, treated_input, thread = 1 ){
registerDoParallel(cores = thread)
testResult <- foreach(i = 1:nrow(control_ip) , .combine = rbind )%dopar%{
x <- t( as.matrix(control_ip[i,]) )
y <- t( as.matrix(control_input[i,]) )
xx <- t( as.matrix(treated_ip[i,]) )
yy <- t( as.matrix(treated_input[i,]) )
xxx = cbind(x,xx)
yyy = cbind(y,yy)
logl1 <- MeTDiff:::.betabinomial.lh(x,y+1)
logl2 <- MeTDiff:::.betabinomial.lh(xx,yy+1)
logl3 <- MeTDiff:::.betabinomial.lh(xxx,yyy+1)
tst <- (logl1$logl+logl2$logl-logl3$logl)*2
pvalues <- 1 - pchisq(tst,2)
log.fc <- log( (sum(xx)+1)/(1+sum(yy)) * (1+sum(y))/(1+sum(x)) )
data.frame( logFC = log.fc, pvalue = pvalues )
}
rm(list=ls(name=foreach:::.foreachGlobals), pos=foreach:::.foreachGlobals)
testResult$fdr <- p.adjust(testResult$pvalue, method = "fdr")
return(testResult)
}
Bltest <- function(control_ip, treated_ip, control_input, treated_input){
control_ip_total <- sum(colSums(control_ip))
control_input_total <- sum(colSums(control_input))
treated_ip_total <- sum(colSums(treated_ip))
treated_input_total <- sum(colSums(treated_input))
tmpResult <- do.call(cbind.data.frame, exomePeak::bltest(rowSums(control_ip), rowSums(control_input),rowSums(treated_ip),rowSums(treated_input),control_ip_total, control_input_total, treated_ip_total, treated_input_total) )
return( data.frame(logFC = tmpResult[,"log.fc"], pvalue = exp(tmpResult[,"log.p"]), fdr = exp(tmpResult$log.fdr) ) )
}In order to compare performance of other method on this data set, we run other three methods on default mode on this dataset.
filteredBins <- rownames(RADAR@ip_adjExpr_filtered)
Metdiff.res <-MeTDiffTest(control_ip = round( RADAR@norm.ip[filteredBins,1:7] ) ,
treated_ip = round( RADAR@norm.ip[filteredBins,8:15] ),
control_input = round( RADAR@norm.input[filteredBins,1:7] ),
treated_input = round( RADAR@norm.input[filteredBins,8:15] ) , thread = 20)
QNB.res <- QNB::qnbtest(control_ip = round( RADAR@norm.ip[filteredBins,1:7] ) ,
treated_ip = round( RADAR@norm.ip[filteredBins,8:15] ),
control_input = round( RADAR@norm.input[filteredBins,1:7] ),
treated_input = round( RADAR@norm.input[filteredBins,8:15] ) ,plot.dispersion = FALSE )
fisher.res <- fisherTest(control_ip = round( RADAR@norm.ip[filteredBins,1:7] ) ,
treated_ip = round( RADAR@norm.ip[filteredBins,8:15] ),
control_input = round( RADAR@norm.input[filteredBins,1:7] ),
treated_input = round( RADAR@norm.input[filteredBins,8:15] ) )
exomePeak.res <- Bltest(control_ip = round( RADAR@norm.ip[filteredBins,1:7] ) ,
treated_ip = round( RADAR@norm.ip[filteredBins,8:15] ),
control_input = round( RADAR@norm.input[filteredBins,1:7] ),
treated_input = round( RADAR@norm.input[filteredBins,8:15] ) )Compare distribution of p value.
pvalues <- data.frame(pvalue = c(RADAR@test.est[,"p_value"],Metdiff.res$pvalue,QNB.res$pvalue,fisher.res$pvalue, exomePeak.res$pvalue ),
method = factor(rep(c("RADAR","MeTDiff","QNB","Fisher","exomePeak"),c(length(RADAR@test.est[,"p_value"]),
length(Metdiff.res$pvalue),
length(QNB.res$pvalue),
length(fisher.res$pvalue),
length(exomePeak.res$pvalue)
)
),levels =c("RADAR","MeTDiff","QNB","Fisher","exomePeak")
)
)
ggplot(pvalues, aes(x = pvalue))+geom_histogram(breaks = seq(0,1,0.04),col=I("black"))+facet_grid(.~method)+theme_bw()+xlab("p-value")+theme( axis.title = element_text(size = 22, face = "bold"),strip.text = element_text(size = 25, face = "bold"),axis.text.x = element_text(size = 18, face = "bold",colour = "black"),axis.text.y = element_text(size = 15, face = "bold",colour = "black",angle = 90),panel.grid = element_blank(), axis.line = element_line(size = 0.7 ,colour = "black"),axis.ticks = element_line(colour = "black"), panel.spacing = unit(0.4, "lines") )+ scale_x_continuous(breaks = seq(0,1,0.2),labels=function(x) sprintf("%.1f", x))+scale_y_continuous(breaks = c(0,25000,50000))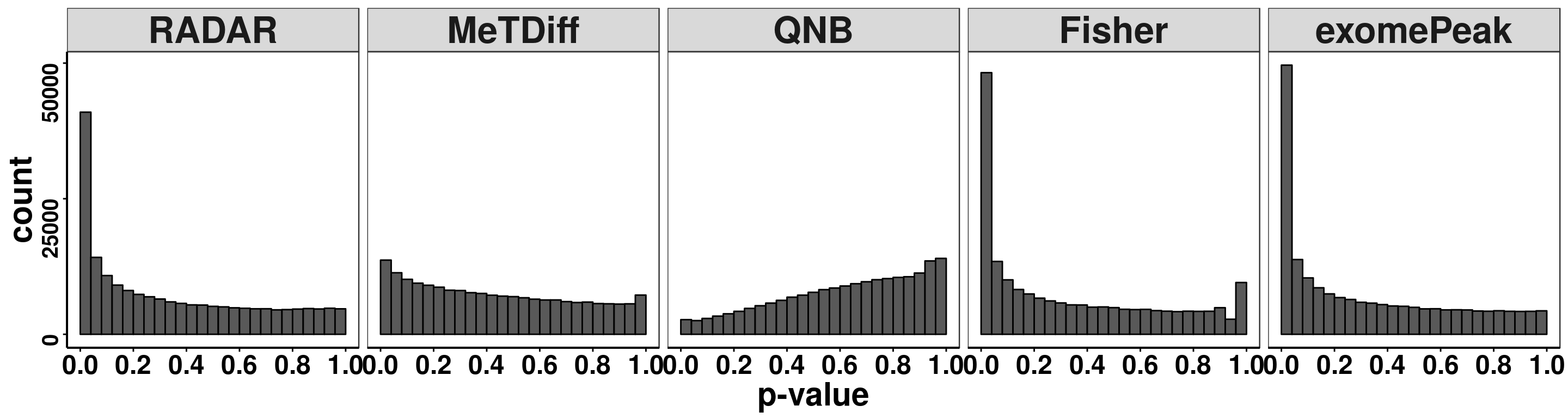
Compare the distibution of beta
par(mfrow = c(1,5))
hist(RADAR@test.est[,"beta"],main = "RADAR",xlab = "beta",breaks = 100,col =rgb(0.2,0.2,0.2,0.5),cex.main = 2.5,cex.axis =2,cex.lab=2)
hist(Metdiff.res$logFC, main = "MeTDiff",xlab = "beta",breaks = 100,col =rgb(0.2,0.2,0.2,0.5),cex.main = 2.5,cex.axis =2,cex.lab=2)
hist(QNB.res$log2.OR,main = "QNB", xlab = "beta",breaks = 100,col =rgb(0.2,0.2,0.2,0.5),cex.main = 2.5,cex.axis =2,cex.lab=2)
hist(fisher.res$logFC, main = "Fisher",xlab = "beta",breaks = 100,col =rgb(0.2,0.2,0.2,0.5),cex.main = 2.5,cex.axis =2,cex.lab=2)
hist(exomePeak.res$logFC, main = "exomePeak",xlab = "beta",breaks = 100,col =rgb(0.2,0.2,0.2,0.5),cex.main = 2.5,cex.axis =2,cex.lab=2)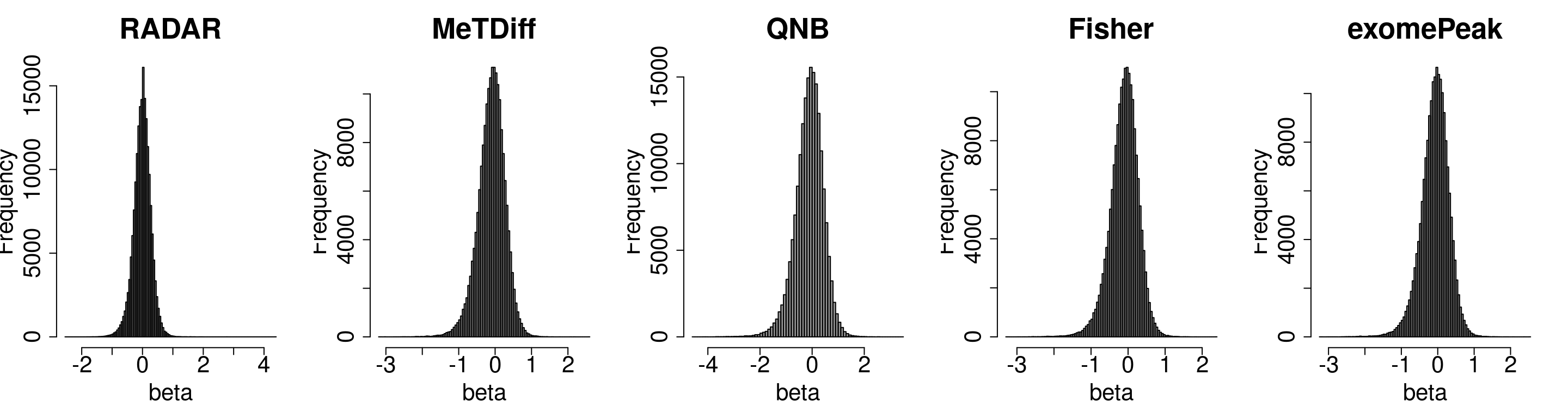
Compare norm by geneSum with norm by position
tmp <- hist(c(RADAR_pos@test.est[,"p_value"]),plot = F)
tmp$counts <- tmp$counts*(nrow(RADAR@ip_adjExpr_filtered)/nrow(RADAR_pos@ip_adjExpr_filtered))
hist(RADAR@test.est[,"p_value"],main = "RADAR",xlab = "p value",col =rgb(1,0,0,0.4),cex.main = 2.5,cex.axis =2,cex.lab=2, ylim = c(0,max(hist(RADAR@test.est[,"p_value"],plot = F)$counts,tmp$counts)) )
plot(tmp,col=rgb(0,0,1,0.5),add = T)
axis(side = 1, lwd = 1,cex.axis =2)
axis(side = 2, lwd = 1,cex.axis =2)
legend("topright", c("By geneSum", "By position"), col=c(rgb(1,0,0,0.4), rgb(0,0,1,0.5)), lwd = 10,bty="n",text.font = 2)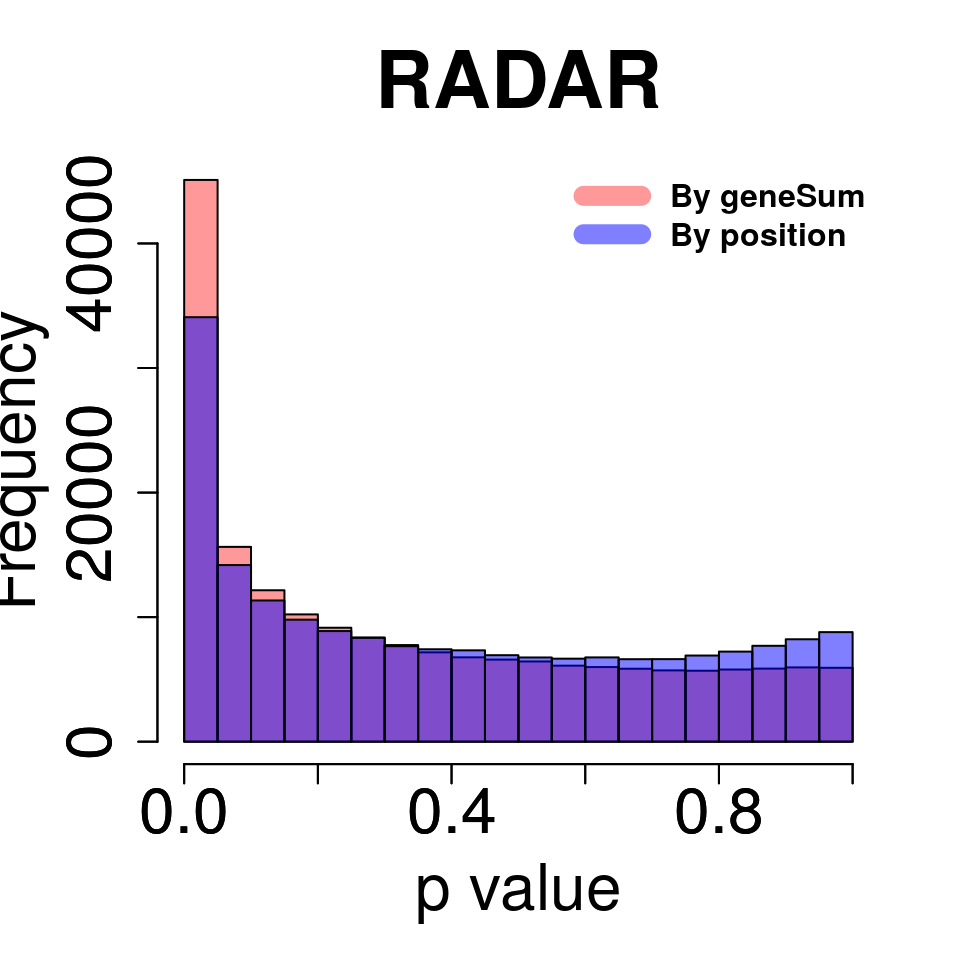
Number of significant bins detected at qvalue < 0.1
sigBins <- apply(cbind("RADAR"=RADAR@test.est[,"p_value"],"MeTDiff"=Metdiff.res$pvalue,"QNB"=QNB.res$pvalue,"Fisher"=fisher.res$pvalue,"exomePeak" = exomePeak.res$pvalue),2, function(x){
length( which( p.adjust(x,method = 'fdr') < 0.1 ) )
})
print(sigBins)## RADAR MeTDiff QNB Fisher exomePeak
## 26613 17 34 38788 40146self-FDR test
Subset samples and run the test.
selfFDR <- NULL
samplenames <- samplenames(RADAR)
set.seed(10)
for(i in 1:15){
## Sample subset
sample_ctl <- sample(x = 1:7, size = 5,replace = F)
sample_case <- 7+sample(x = 1:8, size = 6,replace = F)
sample_id <- c( sample_ctl, sample_case )
if(length(unique(X2[sample_id])) == 1 ){ ## samples all from one batch
## run radar
RADAR_sample <- RADAR
variable(RADAR_sample) <- variable(RADAR)[,c(1,3:4)]
radar_sample <- diffIP_parallel(RADAR_sample, exclude = samplenames[setdiff(1:15,sample_id)], thread = 25 )@test.est
radar_truth <- RADAR@test.est
radar_selfFDR <- sapply(c(0.1,0.05,0.01),function(x){length( which(! which(radar_sample[,"fdr"]<x) %in% which(radar_truth[,"fdr"]<0.1) ) )/length(which(radar_sample[,"fdr"]<x))})
## run MeTDiff
metadiff_sample <-MeTDiffTest(control_ip = round( RADAR@norm.ip[filteredBins,sample_ctl] ) ,
treated_ip = round( RADAR@norm.ip[filteredBins,sample_case] ),
control_input = round( RADAR@norm.input[filteredBins,sample_ctl] ),
treated_input = round( RADAR@norm.input[filteredBins,sample_case] ) , thread = 20)
metdiff_truth <- Metdiff.res
metdiff_selfFDR <- sapply(c(0.1,0.05,0.01),function(x){length( which(! which(metadiff_sample[,"fdr"]<x) %in% which(metdiff_truth[,"fdr"]<0.1) ) )/length(which(metadiff_sample[,"fdr"]<x))})
## run QNB
qnb_sample <- QNB::qnbtest(control_ip = round( RADAR@norm.ip[filteredBins,sample_ctl] ) ,
treated_ip = round( RADAR@norm.ip[filteredBins,sample_case] ),
control_input = round( RADAR@norm.input[filteredBins,sample_ctl] ),
treated_input = round( RADAR@norm.input[filteredBins,sample_case] ) ,plot.dispersion = FALSE )
qnb_truth <- QNB.res
qnb_selfFDR <- sapply(c(0.1,0.05,0.01),function(x){length( which(! which(qnb_sample[,"padj"]<x) %in% which(qnb_truth[,"padj"]<0.1) ) )/length(which(qnb_sample[,"padj"]<x))})
## run Fisher's exact test
fisher_sample <- fisherTest(control_ip = round( RADAR@norm.ip[filteredBins,sample_ctl] ) ,
treated_ip = round( RADAR@norm.ip[filteredBins,sample_case] ),
control_input = round( RADAR@norm.input[filteredBins,sample_ctl] ),
treated_input = round( RADAR@norm.input[filteredBins,sample_case] ) , thread = 20)
fisher_truth <- fisher.res
fisher_selfFDR <- sapply(c(0.1,0.05,0.01),function(x){length( which(! which(fisher_sample[,"fdr"]<x) %in% which(fisher_truth[,"fdr"]<0.1) ) )/length(which(fisher_sample[,"fdr"]<x))})
## run Bltest of the exomePeak
exomePeak_sample <- Bltest(control_ip = round( RADAR@norm.ip[filteredBins,sample_ctl] ) ,
treated_ip = round( RADAR@norm.ip[filteredBins,sample_case] ),
control_input = round( RADAR@norm.input[filteredBins,sample_ctl] ),
treated_input = round( RADAR@norm.input[filteredBins,sample_case] ) )
exomePeak_truth <- exomePeak.res
exomePeak_selfFDR <- sapply(c(0.1,0.05,0.01),function(x){length( which(! which(exomePeak_sample[,"fdr"]<x) %in% which(exomePeak_truth[,"fdr"]<0.1) ) )/length(which(exomePeak_sample[,"fdr"]<x))})
## Summanrize selfFDR
fdr <- rbind( data.frame(self_FDR = radar_selfFDR, fdr_cutoff = c("0.1","0.05","0.01"), method = rep("RADAR",3) ),
data.frame(self_FDR = metdiff_selfFDR, fdr_cutoff = c("0.1","0.05","0.01"), method = rep("MeTDiff",3) ),
data.frame(self_FDR = qnb_selfFDR, fdr_cutoff = c("0.1","0.05","0.01"), method = rep("QNB",3) ),
data.frame(self_FDR = fisher_selfFDR, fdr_cutoff = c("0.1","0.05","0.01"), method = rep("Fisher",3) ),
data.frame(self_FDR = exomePeak_selfFDR, fdr_cutoff = c("0.1","0.05","0.01"), method = rep("exomePeak",3) )
)
selfFDR <- rbind(selfFDR,fdr)
}else if(length(unique(X3[sample_id])) == 1 ){
## run radar
RADAR_sample <- RADAR
variable(RADAR_sample) <- variable(RADAR)[,-c(3)]
radar_sample <- diffIP( RADAR_sample, exclude = samplenames[setdiff(1:15,sample_id)] )@test.est
radar_truth <- RADAR@test.est
radar_selfFDR <- sapply(c(0.1,0.05,0.01),function(x){length( which(! which(radar_sample[,"fdr"]<x) %in% which(radar_truth[,"fdr"]<0.1) ) )/length(which(radar_sample[,"fdr"]<x))})
## run MeTDiff
metadiff_sample <-MeTDiffTest(control_ip = round( RADAR@norm.ip[filteredBins,sample_ctl] ) ,
treated_ip = round( RADAR@norm.ip[filteredBins,sample_case] ),
control_input = round( RADAR@norm.input[filteredBins,sample_ctl] ),
treated_input = round( RADAR@norm.input[filteredBins,sample_case] ) , thread = 20)
metdiff_truth <- Metdiff.res
metdiff_selfFDR <- sapply(c(0.1,0.05,0.01),function(x){length( which(! which(metadiff_sample[,"fdr"]<x) %in% which(metdiff_truth[,"fdr"]<0.1) ) )/length(which(metadiff_sample[,"fdr"]<x))})
## run QNB
qnb_sample <- QNB::qnbtest(control_ip = round( RADAR@norm.ip[filteredBins,sample_ctl] ) ,
treated_ip = round( RADAR@norm.ip[filteredBins,sample_case] ),
control_input = round( RADAR@norm.input[filteredBins,sample_ctl] ),
treated_input = round( RADAR@norm.input[filteredBins,sample_case] ) ,plot.dispersion = FALSE )
qnb_truth <- QNB.res
qnb_selfFDR <- sapply(c(0.1,0.05,0.01),function(x){length( which(! which(qnb_sample[,"padj"]<x) %in% which(qnb_truth[,"padj"]<0.1) ) )/length(which(qnb_sample[,"padj"]<x))})
## run Fisher's exact test
fisher_sample <- fisherTest(control_ip = round( RADAR@norm.ip[filteredBins,sample_ctl] ) ,
treated_ip = round( RADAR@norm.ip[filteredBins,sample_case] ),
control_input = round( RADAR@norm.input[filteredBins,sample_ctl] ),
treated_input = round( RADAR@norm.input[filteredBins,sample_case] ) , thread = 20)
fisher_truth <- fisher.res
fisher_selfFDR <- sapply(c(0.1,0.05,0.01),function(x){length( which(! which(fisher_sample[,"fdr"]<x) %in% which(fisher_truth[,"fdr"]<0.1) ) )/length(which(fisher_sample[,"fdr"]<x))})
## run Bltest of the exomePeak
exomePeak_sample <- Bltest(control_ip = round( RADAR@norm.ip[filteredBins,sample_ctl] ) ,
treated_ip = round( RADAR@norm.ip[filteredBins,sample_case] ),
control_input = round( RADAR@norm.input[filteredBins,sample_ctl] ),
treated_input = round( RADAR@norm.input[filteredBins,sample_case] ) )
exomePeak_truth <- exomePeak.res
exomePeak_selfFDR <- sapply(c(0.1,0.05,0.01),function(x){length( which(! which(exomePeak_sample[,"fdr"]<x) %in% which(exomePeak_truth[,"fdr"]<0.1) ) )/length(which(exomePeak_sample[,"fdr"]<x))})
## Summanrize selfFDR
fdr <- rbind( data.frame(self_FDR = radar_selfFDR, fdr_cutoff = c("0.1","0.05","0.01"), method = rep("RADAR",3) ),
data.frame(self_FDR = metdiff_selfFDR, fdr_cutoff = c("0.1","0.05","0.01"), method = rep("MeTDiff",3) ),
data.frame(self_FDR = qnb_selfFDR, fdr_cutoff = c("0.1","0.05","0.01"), method = rep("QNB",3) ),
data.frame(self_FDR = fisher_selfFDR, fdr_cutoff = c("0.1","0.05","0.01"), method = rep("Fisher",3) ),
data.frame(self_FDR = exomePeak_selfFDR, fdr_cutoff = c("0.1","0.05","0.01"), method = rep("exomePeak",3) )
)
selfFDR <- rbind(selfFDR,fdr)
}else if(length(unique(X4[sample_id])) == 1){
## run radar
RADAR_sample <- RADAR
variable(RADAR_sample) <- variable(RADAR)[,1:3]
radar_sample <- diffIP(RADAR_sample, exclude = samplenames[setdiff(1:15,sample_id)] )@test.est
radar_truth <- RADAR@test.est
radar_selfFDR <- sapply(c(0.1,0.05,0.01),function(x){length( which(! which(radar_sample[,"fdr"]<x) %in% which(radar_truth[,"fdr"]<0.1) ) )/length(which(radar_sample[,"fdr"]<x))})
## run MeTDiff
metadiff_sample <-MeTDiffTest(control_ip = round( RADAR@norm.ip[filteredBins,sample_ctl] ) ,
treated_ip = round( RADAR@norm.ip[filteredBins,sample_case] ),
control_input = round( RADAR@norm.input[filteredBins,sample_ctl] ),
treated_input = round( RADAR@norm.input[filteredBins,sample_case] ) , thread = 20)
metdiff_truth <- Metdiff.res
metdiff_selfFDR <- sapply(c(0.1,0.05,0.01),function(x){length( which(! which(metadiff_sample[,"fdr"]<x) %in% which(metdiff_truth[,"fdr"]<0.1) ) )/length(which(metadiff_sample[,"fdr"]<x))})
## run QNB
qnb_sample <- QNB::qnbtest(control_ip = round( RADAR@norm.ip[filteredBins,sample_ctl] ) ,
treated_ip = round( RADAR@norm.ip[filteredBins,sample_case] ),
control_input = round( RADAR@norm.input[filteredBins,sample_ctl] ),
treated_input = round( RADAR@norm.input[filteredBins,sample_case] ) ,plot.dispersion = FALSE )
qnb_truth <- QNB.res
qnb_selfFDR <- sapply(c(0.1,0.05,0.01),function(x){length( which(! which(qnb_sample[,"padj"]<x) %in% which(qnb_truth[,"padj"]<0.1) ) )/length(which(qnb_sample[,"padj"]<x))})
## run Fisher's exact test
fisher_sample <- fisherTest(control_ip = round( RADAR@norm.ip[filteredBins,sample_ctl] ) ,
treated_ip = round( RADAR@norm.ip[filteredBins,sample_case] ),
control_input = round( RADAR@norm.input[filteredBins,sample_ctl] ),
treated_input = round( RADAR@norm.input[filteredBins,sample_case] ) , thread = 20)
fisher_truth <- fisher.res
fisher_selfFDR <- sapply(c(0.1,0.05,0.01),function(x){length( which(! which(fisher_sample[,"fdr"]<x) %in% which(fisher_truth[,"fdr"]<0.1) ) )/length(which(fisher_sample[,"fdr"]<x))})
## run Bltest of the exomePeak
exomePeak_sample <- Bltest(control_ip = round( RADAR@norm.ip[filteredBins,sample_ctl] ) ,
treated_ip = round( RADAR@norm.ip[filteredBins,sample_case] ),
control_input = round( RADAR@norm.input[filteredBins,sample_ctl] ),
treated_input = round( RADAR@norm.input[filteredBins,sample_case] ) )
exomePeak_truth <- exomePeak.res
exomePeak_selfFDR <- sapply(c(0.1,0.05,0.01),function(x){length( which(! which(exomePeak_sample[,"fdr"]<x) %in% which(exomePeak_truth[,"fdr"]<0.1) ) )/length(which(exomePeak_sample[,"fdr"]<x))})
## Summanrize selfFDR
fdr <- rbind( data.frame(self_FDR = radar_selfFDR, fdr_cutoff = c("0.1","0.05","0.01"), method = rep("RADAR",3) ),
data.frame(self_FDR = metdiff_selfFDR, fdr_cutoff = c("0.1","0.05","0.01"), method = rep("MeTDiff",3) ),
data.frame(self_FDR = qnb_selfFDR, fdr_cutoff = c("0.1","0.05","0.01"), method = rep("QNB",3) ),
data.frame(self_FDR = fisher_selfFDR, fdr_cutoff = c("0.1","0.05","0.01"), method = rep("Fisher",3) ),
data.frame(self_FDR = exomePeak_selfFDR, fdr_cutoff = c("0.1","0.05","0.01"), method = rep("exomePeak",3) )
)
selfFDR <- rbind(selfFDR,fdr)
}else{
## run radar
RADAR_sample <- RADAR
radar_sample <- diffIP( RADAR_sample, exclude = samplenames[setdiff(1:15,sample_id)] )@test.est
radar_truth <- RADAR@test.est
radar_selfFDR <- sapply(c(0.1,0.05,0.01),function(x){length( which(! which(radar_sample[,"fdr"]<x) %in% which(radar_truth[,"fdr"]<0.1) ) )/length(which(radar_sample[,"fdr"]<x))})
## run MeTDiff
metadiff_sample <-MeTDiffTest(control_ip = round( RADAR@norm.ip[filteredBins,sample_ctl] ) ,
treated_ip = round( RADAR@norm.ip[filteredBins,sample_case] ),
control_input = round( RADAR@norm.input[filteredBins,sample_ctl] ),
treated_input = round( RADAR@norm.input[filteredBins,sample_case] ) , thread = 20)
metdiff_truth <- Metdiff.res
metdiff_selfFDR <- sapply(c(0.1,0.05,0.01),function(x){length( which(! which(metadiff_sample[,"fdr"]<x) %in% which(metdiff_truth[,"fdr"]<0.1) ) )/length(which(metadiff_sample[,"fdr"]<x))})
## run QNB
qnb_sample <- QNB::qnbtest(control_ip = round( RADAR@norm.ip[filteredBins,sample_ctl] ) ,
treated_ip = round( RADAR@norm.ip[filteredBins,sample_case] ),
control_input = round( RADAR@norm.input[filteredBins,sample_ctl] ),
treated_input = round( RADAR@norm.input[filteredBins,sample_case] ) ,plot.dispersion = FALSE )
qnb_truth <- QNB.res
qnb_selfFDR <- sapply(c(0.1,0.05,0.01),function(x){length( which(! which(qnb_sample[,"padj"]<x) %in% which(qnb_truth[,"padj"]<0.1) ) )/length(which(qnb_sample[,"padj"]<x))})
## run Fisher's exact test
fisher_sample <- fisherTest(control_ip = round( RADAR@norm.ip[filteredBins,sample_ctl] ) ,
treated_ip = round( RADAR@norm.ip[filteredBins,sample_case] ),
control_input = round( RADAR@norm.input[filteredBins,sample_ctl] ),
treated_input = round( RADAR@norm.input[filteredBins,sample_case] ), thread = 20 )
fisher_truth <- fisher.res
fisher_selfFDR <- sapply(c(0.1,0.05,0.01),function(x){length( which(! which(fisher_sample[,"fdr"]<x) %in% which(fisher_truth[,"fdr"]<0.1) ) )/length(which(fisher_sample[,"fdr"]<x))})
## run Bltest of the exomePeak
exomePeak_sample <- Bltest(control_ip = round( RADAR@norm.ip[filteredBins,sample_ctl] ) ,
treated_ip = round( RADAR@norm.ip[filteredBins,sample_case] ),
control_input = round( RADAR@norm.input[filteredBins,sample_ctl] ),
treated_input = round( RADAR@norm.input[filteredBins,sample_case] ) )
exomePeak_truth <- exomePeak.res
exomePeak_selfFDR <- sapply(c(0.1,0.05,0.01),function(x){length( which(! which(exomePeak_sample[,"fdr"]<x) %in% which(exomePeak_truth[,"fdr"]<0.1) ) )/length(which(exomePeak_sample[,"fdr"]<x))})
## Summanrize selfFDR
fdr <- rbind( data.frame(self_FDR = radar_selfFDR, fdr_cutoff = c("0.1","0.05","0.01"), method = rep("RADAR",3) ),
data.frame(self_FDR = metdiff_selfFDR, fdr_cutoff = c("0.1","0.05","0.01"), method = rep("MeTDiff",3) ),
data.frame(self_FDR = qnb_selfFDR, fdr_cutoff = c("0.1","0.05","0.01"), method = rep("QNB",3) ),
data.frame(self_FDR = fisher_selfFDR, fdr_cutoff = c("0.1","0.05","0.01"), method = rep("Fisher",3) ),
data.frame(self_FDR = exomePeak_selfFDR, fdr_cutoff = c("0.1","0.05","0.01"), method = rep("exomePeak",3) )
)
selfFDR <- rbind(selfFDR,fdr)
}
}
selfFDR$fdr_cutoff <- factor(selfFDR$fdr_cutoff,levels = c("0.1","0.05","0.01"))
save(selfFDR, file = "~/Tools/RADARmannual/data/T2D_selfFDR.RData")load( "~/Tools/RADARmannual/data/T2D_selfFDR.RData")
ggplot(data = selfFDR, aes(x=method, y= self_FDR, colour = fdr_cutoff))+geom_boxplot()+theme_bw() +xlab("Method")+ylab("Self-FDR")+ theme(panel.border = element_blank(), panel.grid.major = element_blank(),
panel.grid.minor = element_blank(), axis.line = element_line(colour = "black",size = 1),
axis.title.x=element_text(size=20, face="bold", hjust=0.5),
axis.title.y=element_text(size=20, face="bold", vjust=0.4, angle=90),
legend.title=element_text(size = 15,face = "bold"),legend.text = element_text(size = 18, face = "bold",family = "arial"),
axis.text = element_text(size = 15,face = "bold",colour = "black") , axis.ticks = element_line(colour = "black",size = 1) )+scale_color_discrete(name = "FDR\ncutoff")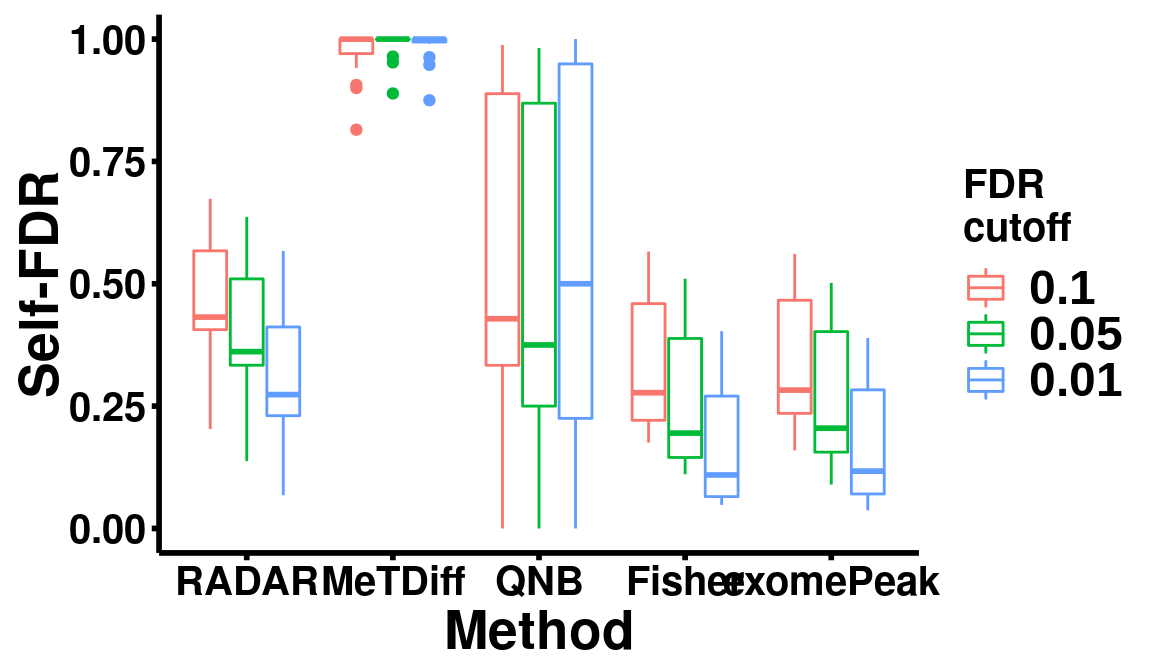
selfFDR_pos <- NULL
samplenames <- RADAR@samplenames
set.seed(3)
for(i in 1:15){
## Sample subset
sample_id <- c(sample(x = 1:7, size = 5,replace = F),7+sample(x = 1:8, size = 6,replace = F) )
Truth_id <- setdiff(1:12,sample_id)
if(length(unique(X2[sample_id])) == 1 ){ ## samples all from one batch
## run radar
RADAR_sample <- RADAR_pos
variable(RADAR_sample) <- variable(RADAR_pos)[,c(1,3:4)]
radar_sample <- diffIP_parallel(RADAR_sample, exclude = samplenames[setdiff(1:15,sample_id)], thread = 20 )@test.est
radar_truth <- RADAR_pos@test.est
radar_selfFDR <- sapply(c(0.1,0.05,0.01),function(x){length( which(! which(radar_sample[,"fdr"]<x) %in% which(radar_truth[,"fdr"]<0.1) ) )/length(which(radar_sample[,"fdr"]<x))})
## Summanrize selfFDR
fdr <- data.frame(self_FDR = radar_selfFDR, fdr_cutoff = c("0.1","0.05","0.01"), method = rep("RADAR_pos",3) )
selfFDR_pos <- rbind(selfFDR_pos,fdr)
}else if(length(unique(X3[sample_id])) == 1 ){
## run radar
RADAR_sample <- RADAR_pos
variable(RADAR_sample) <- variable(RADAR_pos)[,-c(3)]
radar_sample <- diffIP_parallel(RADAR_sample, exclude = samplenames[setdiff(1:15,sample_id)], thread = 20 )@test.est
radar_truth <- RADAR_pos@test.est
radar_selfFDR <- sapply(c(0.1,0.05,0.01),function(x){length( which(! which(radar_sample[,"fdr"]<x) %in% which(radar_truth[,"fdr"]<0.1) ) )/length(which(radar_sample[,"fdr"]<x))})
## Summanrize selfFDR
fdr <- data.frame(self_FDR = radar_selfFDR, fdr_cutoff = c("0.1","0.05","0.01"), method = rep("RADAR_pos",3) )
selfFDR_pos <- rbind(selfFDR_pos,fdr)
}else if(length(unique(X4[sample_id])) == 1){
## run radar
RADAR_sample <- RADAR_pos
variable(RADAR_sample) <- variable(RADAR_pos)[,1:3]
radar_sample <- diffIP_parallel(RADAR_sample, exclude = samplenames[setdiff(1:15,sample_id)], thread = 20 )@test.est
radar_truth <- RADAR_pos@test.est
radar_selfFDR <- sapply(c(0.1,0.05,0.01),function(x){length( which(! which(radar_sample[,"fdr"]<x) %in% which(radar_truth[,"fdr"]<0.1) ) )/length(which(radar_sample[,"fdr"]<x))})
## Summanrize selfFDR
fdr <- data.frame(self_FDR = radar_selfFDR, fdr_cutoff = c("0.1","0.05","0.01"), method = rep("RADAR_pos",3) )
selfFDR_pos <- rbind(selfFDR_pos,fdr)
}else{
## run radar
RADAR_sample <- RADAR_pos
radar_sample <- diffIP_parallel(RADAR_sample, exclude = samplenames[setdiff(1:15,sample_id)], thread = 20 )@test.est
radar_truth <- RADAR_pos@test.est
radar_selfFDR <- sapply(c(0.1,0.05,0.01),function(x){length( which(! which(radar_sample[,"fdr"]<x) %in% which(radar_truth[,"fdr"]<0.1) ) )/length(which(radar_sample[,"fdr"]<x))})
## Summanrize selfFDR
fdr <- data.frame(self_FDR = radar_selfFDR, fdr_cutoff = c("0.1","0.05","0.01"), method = rep("RADAR_pos",3) )
selfFDR_pos <- rbind(selfFDR_pos,fdr)
}
}
selfFDR_pos$fdr_cutoff <- factor(selfFDR_pos$fdr_cutoff,levels = c("0.1","0.05","0.01"))
save(selfFDR_pos, file = "~/Tools/RADARmannual/data/T2D_selfFDR_pos.RData")Self-FDR norm by local bin read count
load( "~/Tools/RADARmannual/data/T2D_selfFDR_pos.RData")
selfFDR_all <- rbind(selfFDR, selfFDR_pos)
selfFDR_all$method <- factor(selfFDR_all$method,levels = c( "RADAR","RADAR_pos","MeTDiff","QNB","Fisher","exomePeak" ))
ggplot(data = selfFDR_all, aes(x=method, y= self_FDR, colour = fdr_cutoff))+geom_boxplot()+theme_bw() +xlab("Method")+ylab("Self-FDR")+ theme( panel.grid.major = element_blank(),
axis.line = element_line(colour = "black",size = 1),
axis.title.x=element_text(size=20, face="bold", hjust=0.5,family = "arial"),
axis.title.y=element_text(size=20, face="bold", vjust=0.4, angle=90,family = "arial"),
legend.title=element_text(size = 15,face = "bold"),legend.text = element_text(size = 18, face = "bold",family = "arial"),
axis.text = element_text(size = 18,face = "bold",family = "arial",colour = "black") ,strip.text.x = element_text(size = 20,face = "bold") )+scale_color_discrete(name = "FDR\ncutoff")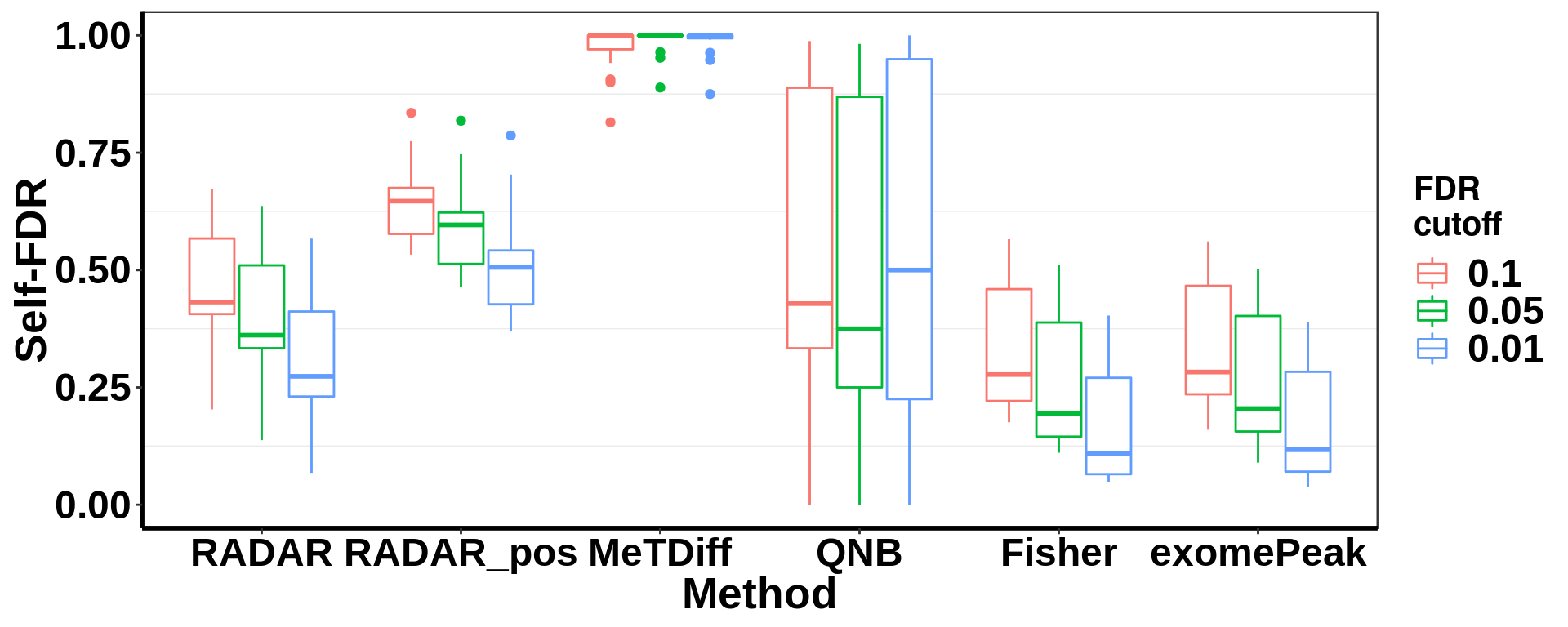
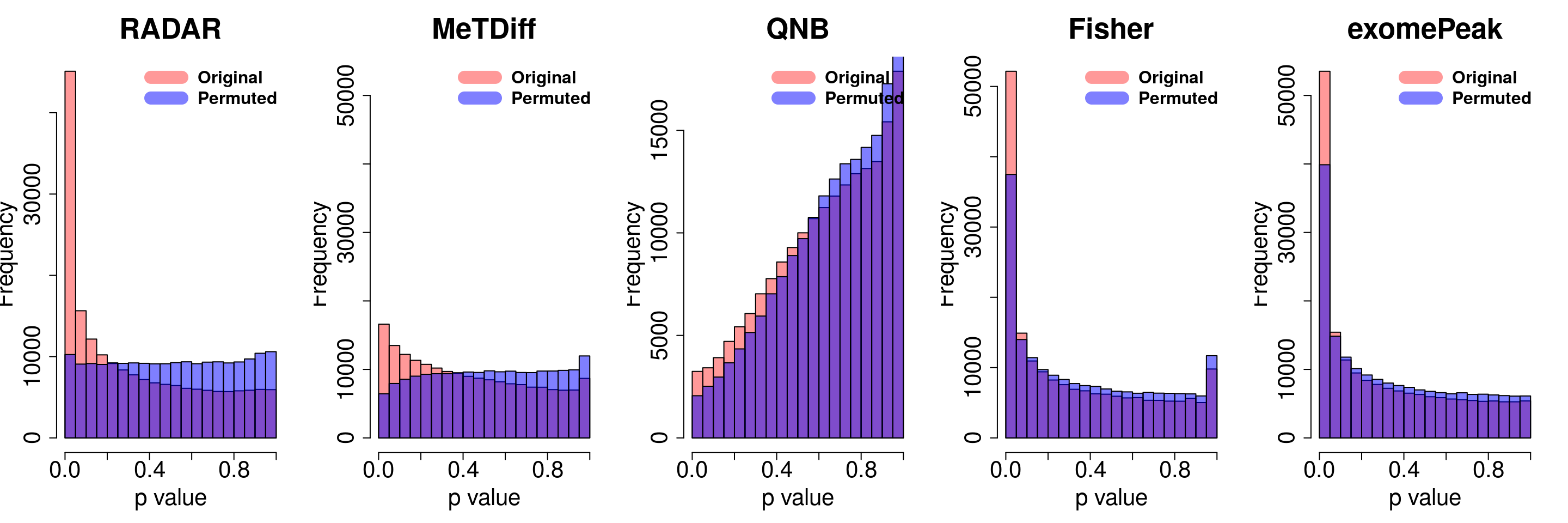
Permution
radar_permuted_P <-NULL
metdiff_permuted_P <- NULL
qnb_permuted_P <- NULL
fisher_permuted_P <- NULL
exomePeak_permute_P <- NULL
set.seed(51)
for(i in 1:15){
permute_X <- variable(RADAR)[,1]
newT2Did <- foreach(bat = unique(batch), .combine = c )%do%{
tmp_id <- which(batch %in% bat)
tmp_X <- variable(RADAR)[tmp_id,1]
return ( c(sample(x = tmp_id[which(tmp_X == "Ctl")], size = round(length(which(tmp_X == "Ctl"))/2) ),
sample(x = tmp_id[which(tmp_X == "T2D")], size = round(length(which(tmp_X == "T2D"))/2) )) )
}
permute_X[newT2Did] <- "T2D"
permute_X[setdiff(1:15,newT2Did)] <- "Ctl"
RADAR_permute <- RADAR
variable(RADAR_permute) <- cbind( condition=permute_X, variable(RADAR)[2:3] )
if ( all( permute_X == unlist(variable(RADAR)[,1]) ) | all( permute_X == c(rep("T2D",8),rep("Ctl",7))) ){
cat("Duplicated with original design...\n")
}else{
radar_permute <- diffIP_parallel( RADAR_permute , thread = 20 )@test.est[,"p_value"]
metdiff_permute <-MeTDiffTest( control_ip = round( RADAR@norm.ip[filteredBins ,(which(permute_X == "Ctl" ) )] ),
treated_ip = round( RADAR@norm.ip[filteredBins,(which(permute_X == "T2D" ) )] ),
control_input = round( RADAR@norm.input[filteredBins,which(permute_X == "Ctl" )] ),
treated_input = round( RADAR@norm.input[filteredBins,which(permute_X == "T2D" )] ) ,
thread = 25)$pvalue
qnb_permute <- QNB::qnbtest(control_ip = round( RADAR@norm.ip[filteredBins ,(which(permute_X == "Ctl" ) )] ),
treated_ip = round( RADAR@norm.ip[filteredBins,(which(permute_X == "T2D" ) )] ),
control_input = round( RADAR@norm.input[filteredBins,which(permute_X == "Ctl" )] ),
treated_input = round( RADAR@norm.input[filteredBins,which(permute_X == "T2D" )] ),
plot.dispersion = FALSE)$pvalue
fisher_permute <- fisherTest( control_ip = round( RADAR@norm.ip[filteredBins ,(which(permute_X == "Ctl" ) )] ),
treated_ip = round( RADAR@norm.ip[filteredBins,(which(permute_X == "T2D" ) )] ),
control_input = round( RADAR@norm.input[filteredBins,which(permute_X == "Ctl" )] ),
treated_input = round( RADAR@norm.input[filteredBins,which(permute_X == "T2D" )] ) ,
thread = 25
)$pvalue
exomePeak_permute <- Bltest( control_ip = round( RADAR@norm.ip[filteredBins ,(which(permute_X == "Ctl" ) )] ),
treated_ip = round( RADAR@norm.ip[filteredBins,(which(permute_X == "T2D" ) )] ),
control_input = round( RADAR@norm.input[filteredBins,which(permute_X == "Ctl" )] ),
treated_input = round( RADAR@norm.input[filteredBins,which(permute_X == "T2D" )] )
)$pvalue
radar_permuted_P <- cbind(radar_permuted_P, radar_permute)
metdiff_permuted_P <- cbind(metdiff_permuted_P, metdiff_permute)
qnb_permuted_P <- cbind(qnb_permuted_P,qnb_permute)
fisher_permuted_P <- cbind(fisher_permuted_P, fisher_permute)
exomePeak_permute_P <- cbind(exomePeak_permute_P, exomePeak_permute)
}
}for( i in 1:ncol(radar_permuted_P)){
par(mfrow=c(1,5))
hist(RADAR@test.est[,"p_value"], col =rgb(1,0,0,0.4),main = "RADAR", xlab = "p value",cex.main = 2.5,cex.axis =2,cex.lab=2)
hist(radar_permuted_P[,i],col=rgb(0,0,1,0.5),add = T)
legend("topright", c("Original", "Permuted"), col=c(rgb(1,0,0,0.4), rgb(0,0,1,0.5)), lwd = 12,bty="n",text.font = 2, cex = 1.5)
hist(Metdiff.res$pvalue, col =rgb(1,0,0,0.4),main = "MeTDiff",xlab = "p value",cex.main = 2.5,cex.axis =2,cex.lab=2, ylim =c(0,max(hist(exomePeak.res$pvalue,plot = F)$counts) ) )
hist(metdiff_permuted_P[,i],col=rgb(0,0,1,0.5),add = T)
legend("topright", c("Original", "Permuted"), col=c(rgb(1,0,0,0.4), rgb(0,0,1,0.5)), lwd = 12,bty="n",text.font = 2, cex = 1.5)
hist(QNB.res$pvalue, col =rgb(1,0,0,0.4),main = "QNB",xlab = "p value",cex.main = 2.5,cex.axis =2,cex.lab=2)
hist(qnb_permuted_P[,i],col=rgb(0,0,1,0.5),add = T)
legend("topright", c("Original", "Permuted"), col=c(rgb(1,0,0,0.4), rgb(0,0,1,0.5)), lwd = 12,bty="n",text.font = 2, cex = 1.5)
hist(fisher.res$pvalue, col =rgb(1,0,0,0.4),main = "Fisher",xlab = "p value",cex.main = 2.5,cex.axis =2,cex.lab=2)
hist(fisher_permuted_P[,i],col=rgb(0,0,1,0.5),add = T)
legend("topright", c("Original", "Permuted"), col=c(rgb(1,0,0,0.4), rgb(0,0,1,0.5)), lwd = 12,bty="n",text.font = 2, cex = 1.5)
hist(exomePeak.res$pvalue, col =rgb(1,0,0,0.4),main = "exomePeak",xlab = "p value",cex.main = 2.5,cex.axis =2,cex.lab=2)
hist(exomePeak_permute_P[,i],col=rgb(0,0,1,0.5),add = T)
legend("topright", c("Original", "Permuted"), col=c(rgb(1,0,0,0.4), rgb(0,0,1,0.5)), lwd = 12,bty="n",text.font = 2, cex = 1.5)
}
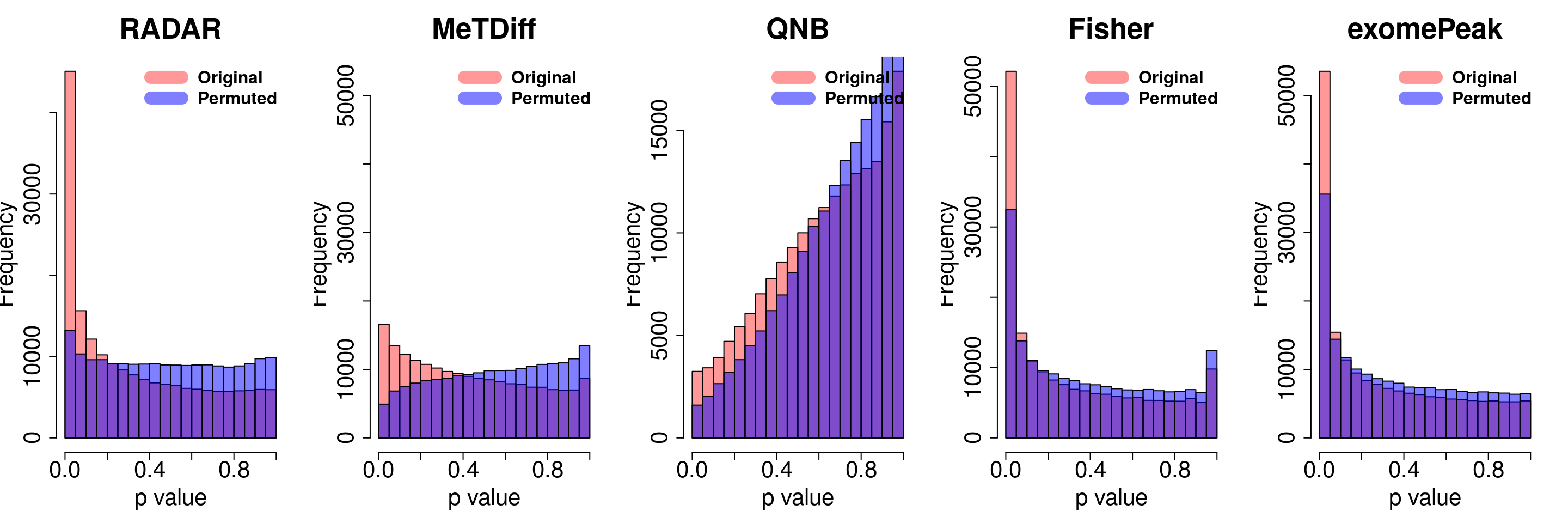
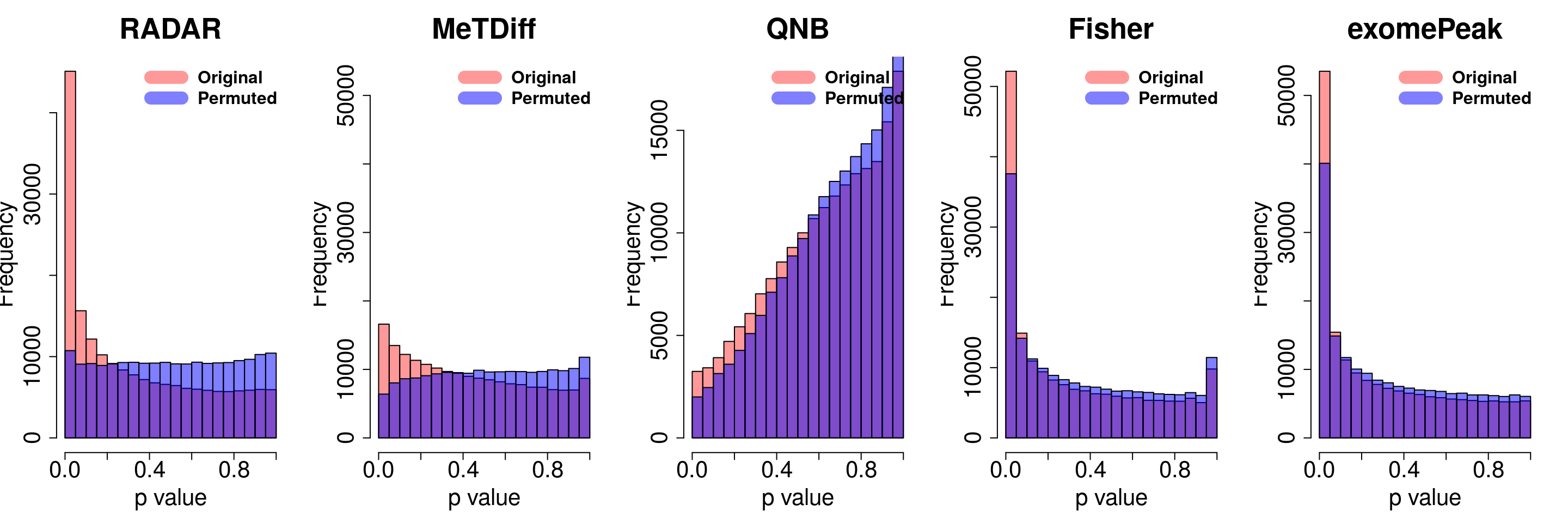
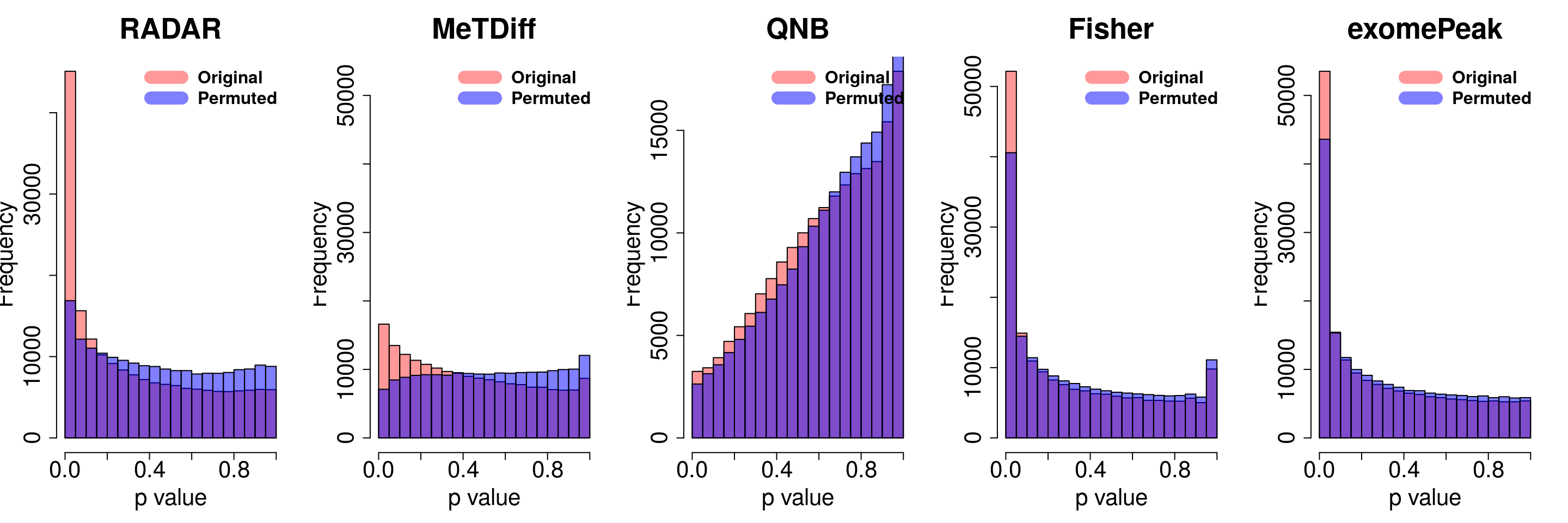
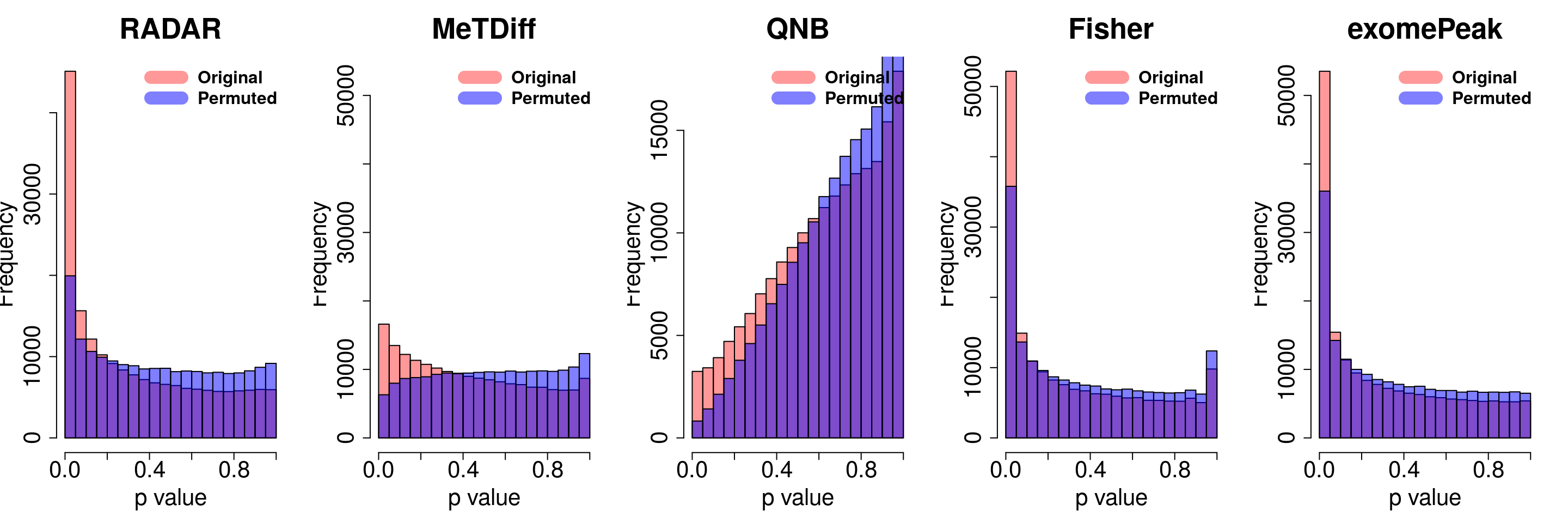
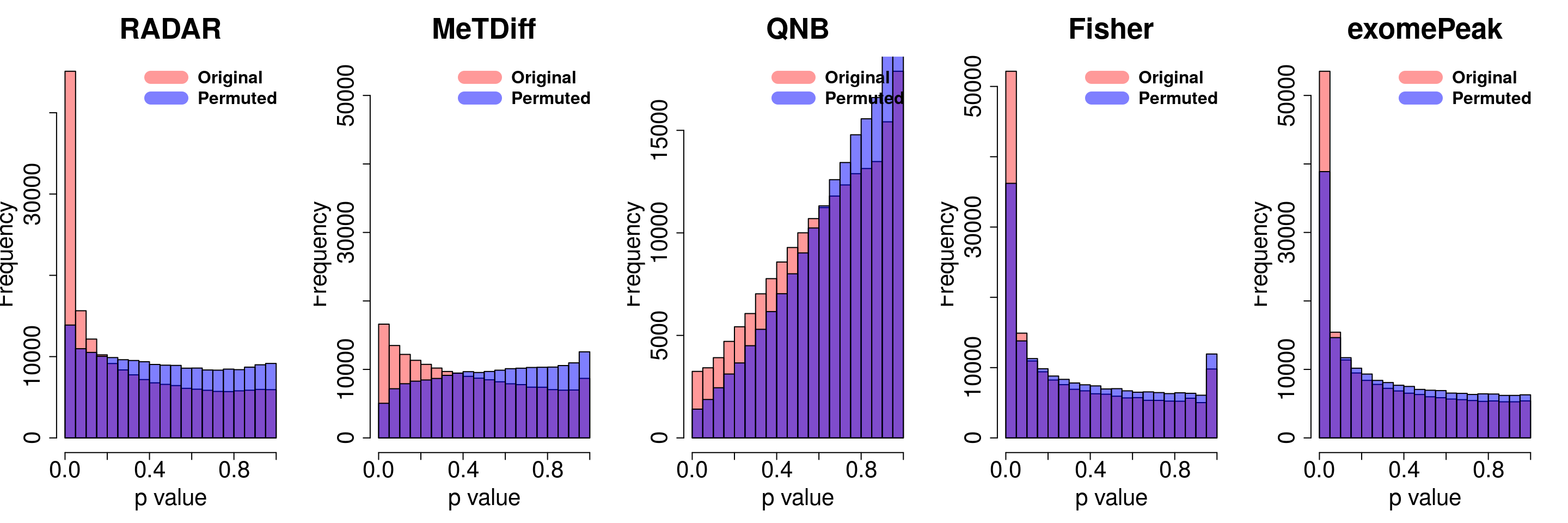
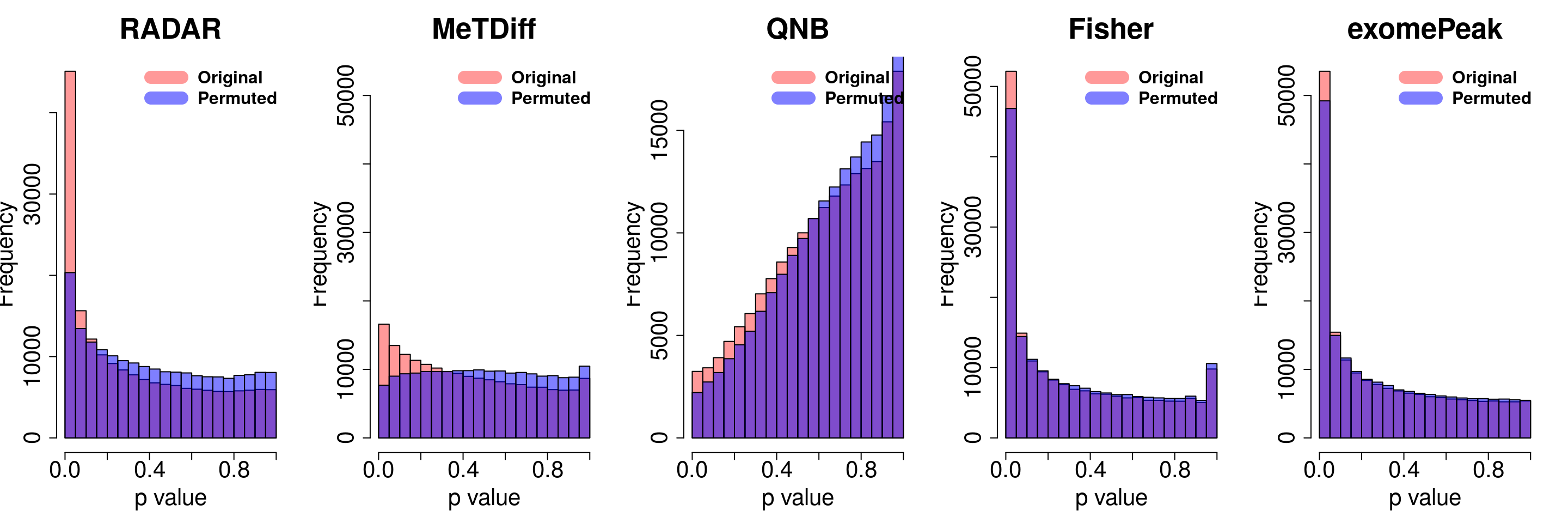
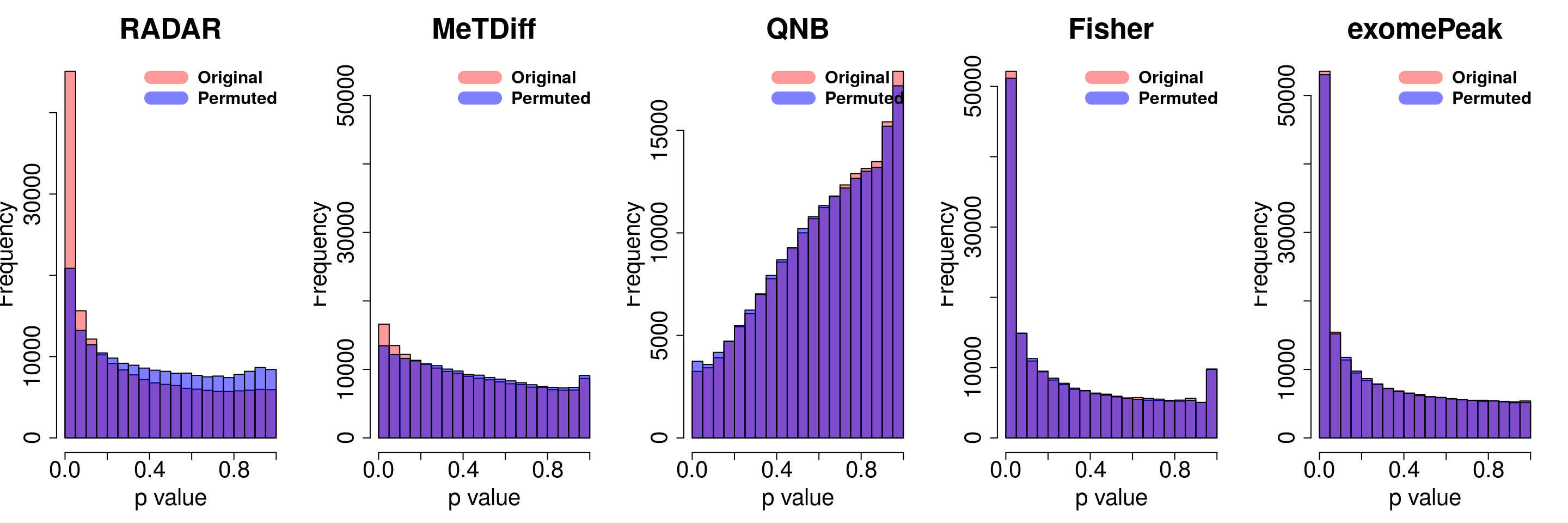
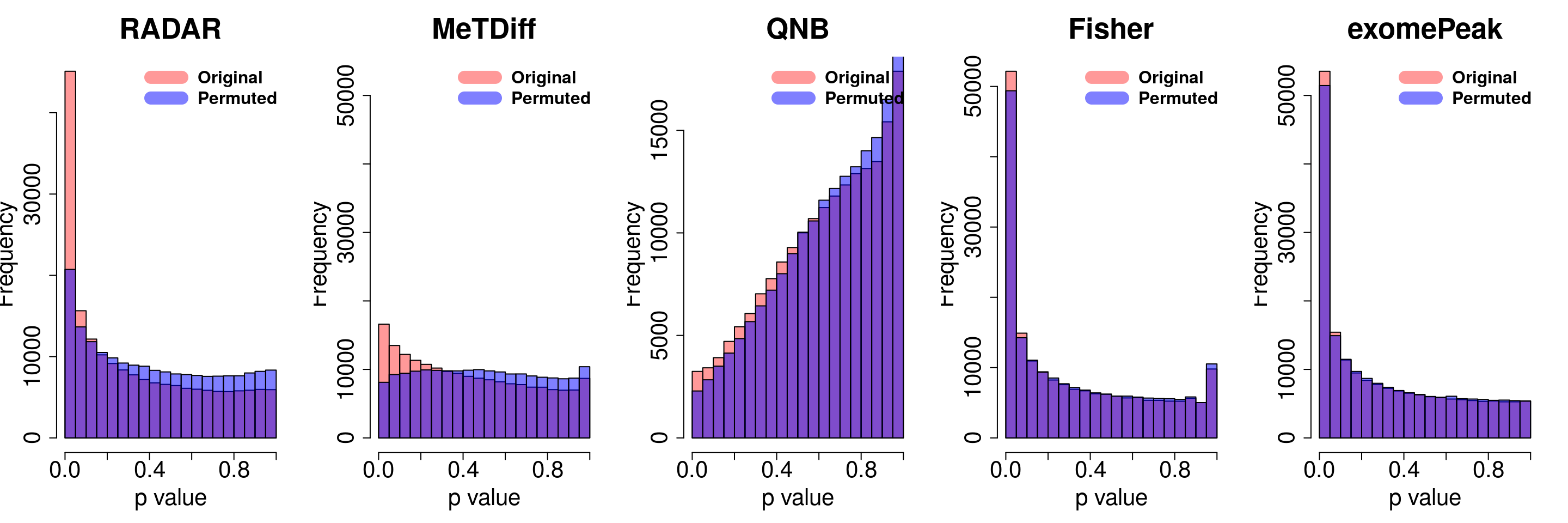
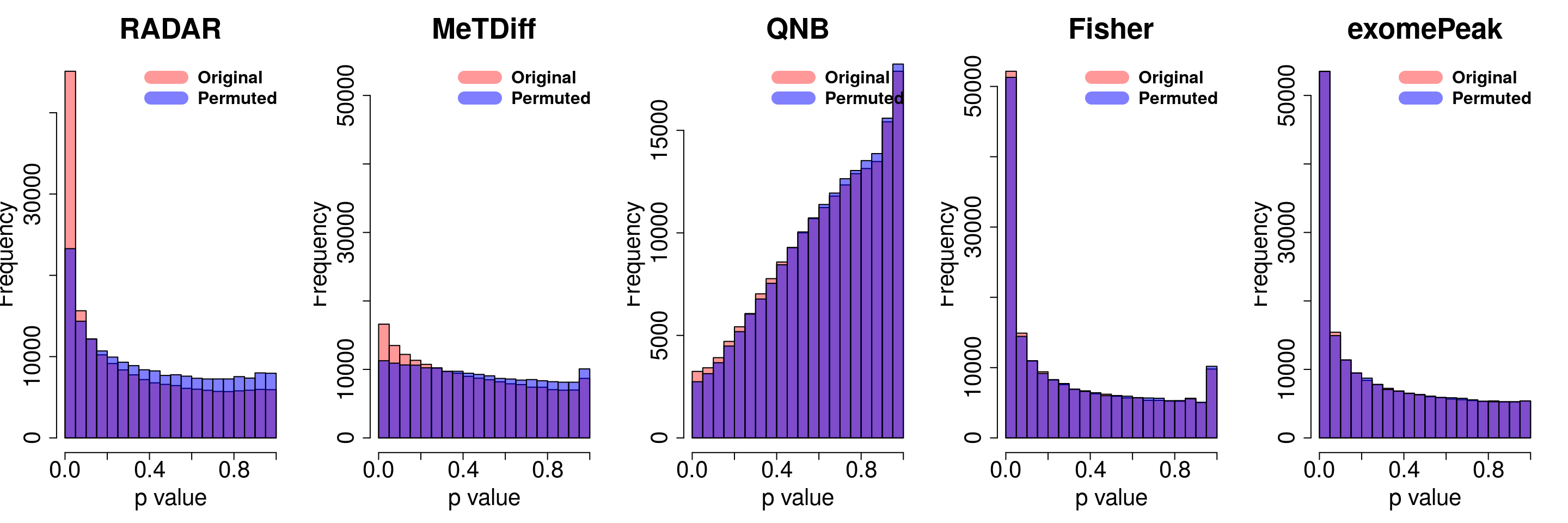
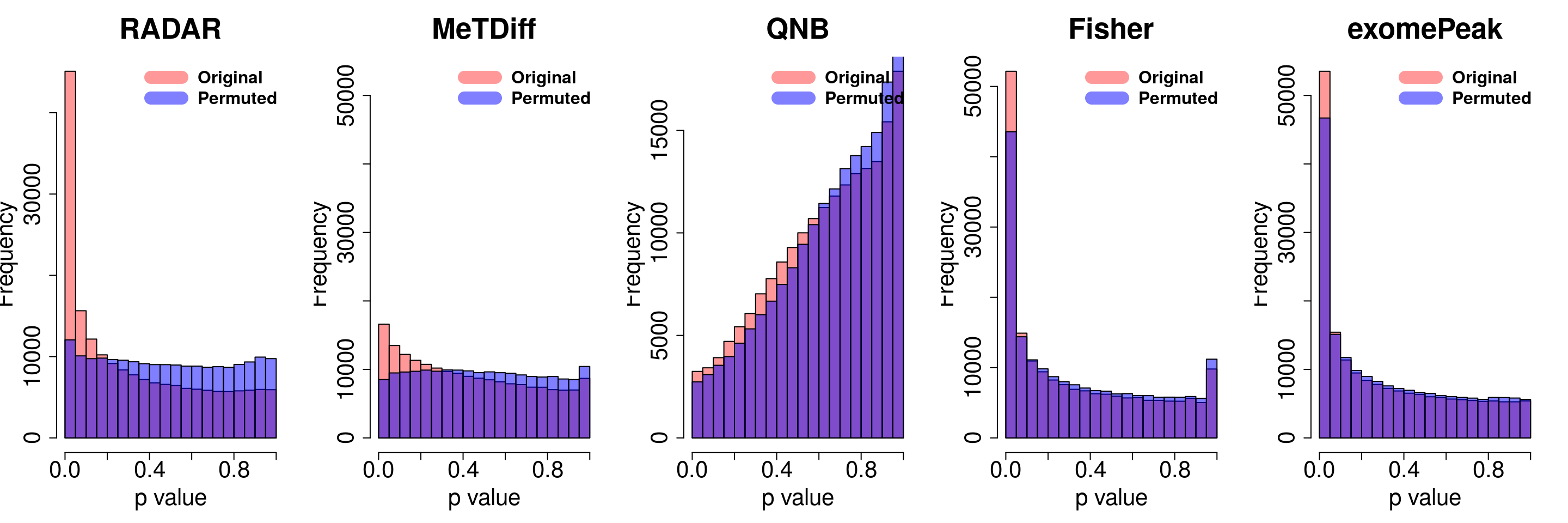
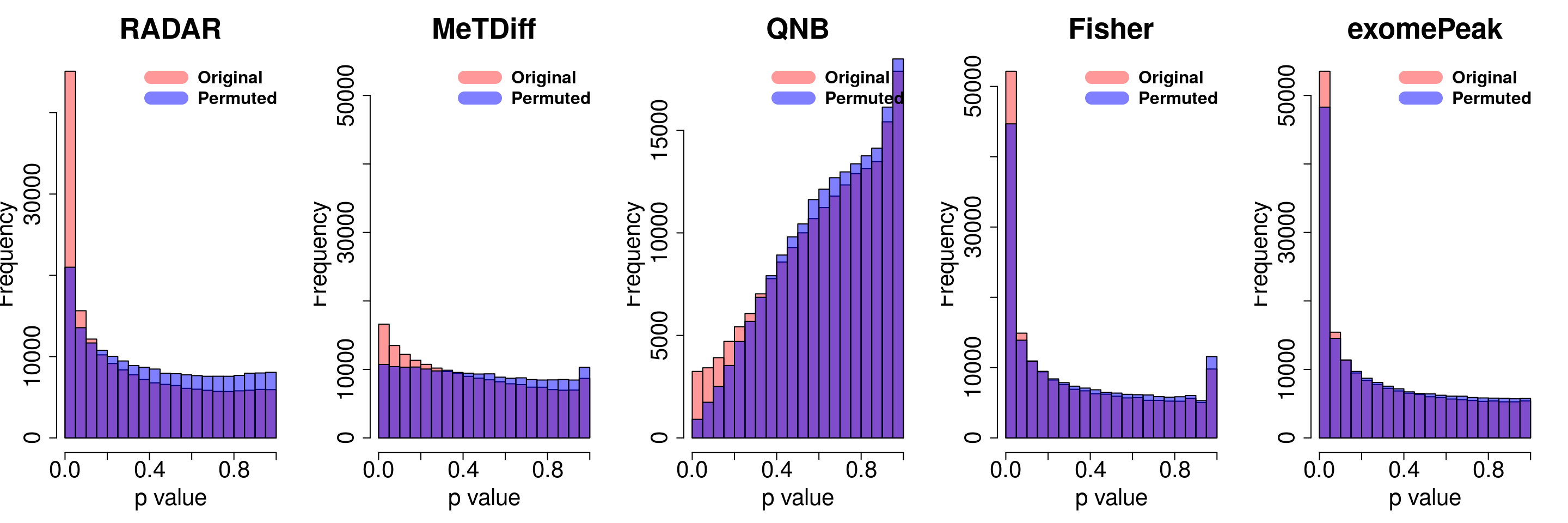
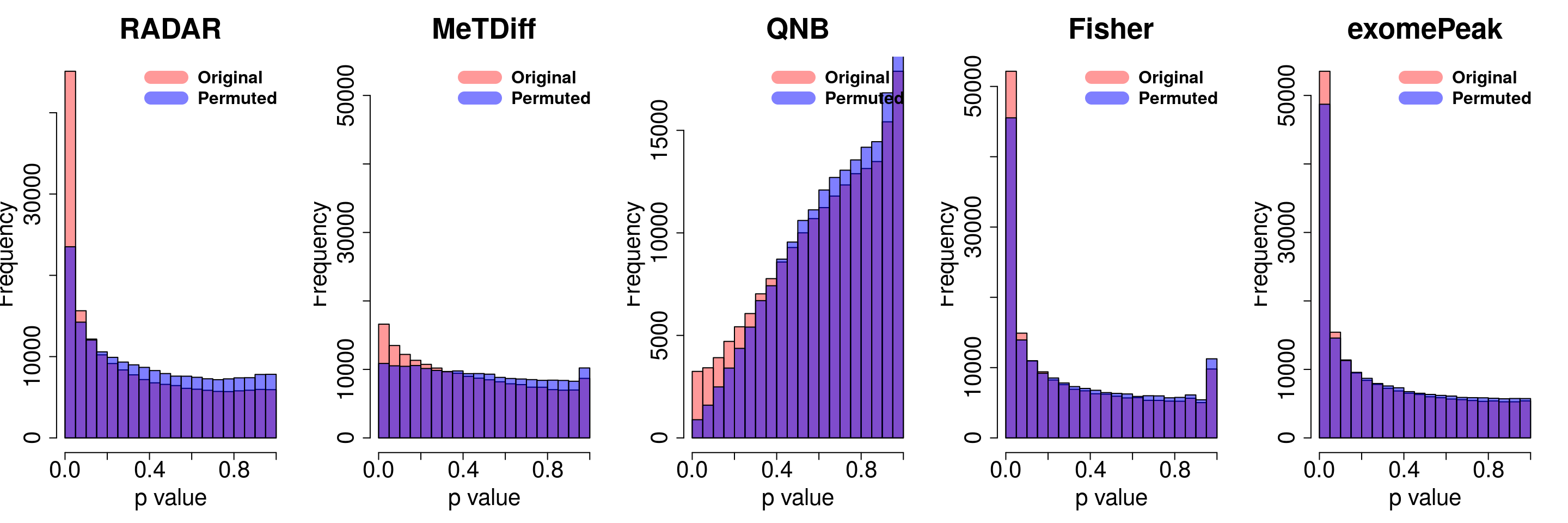
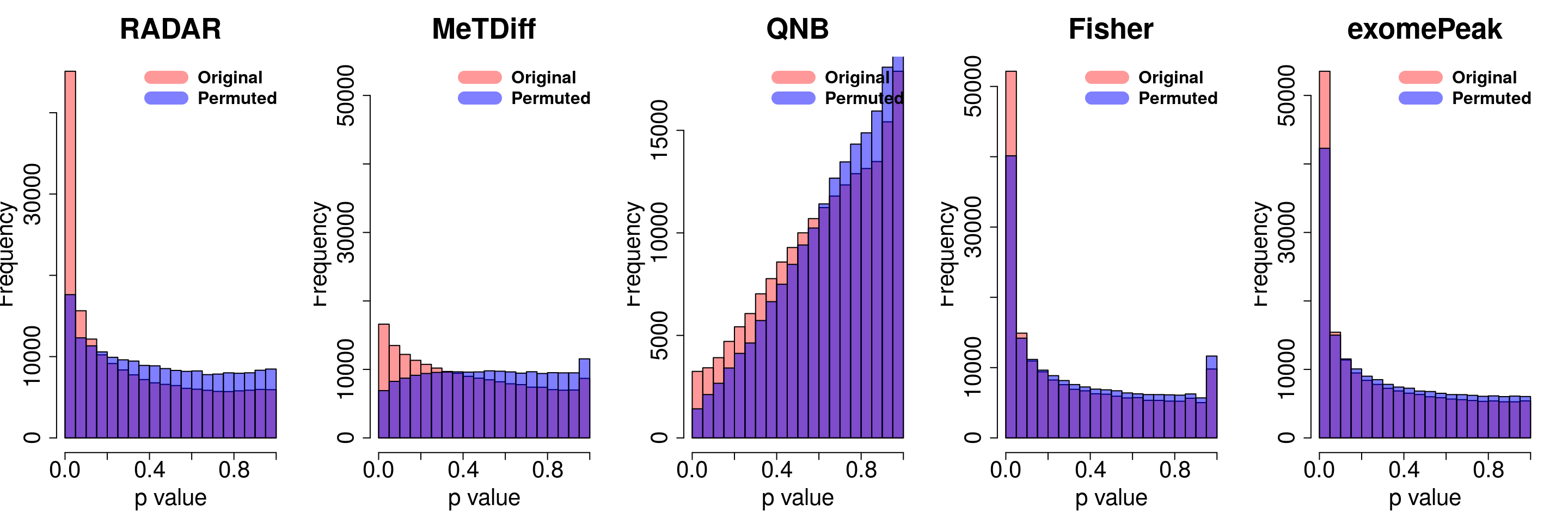
combine all sets of permuted p value in one
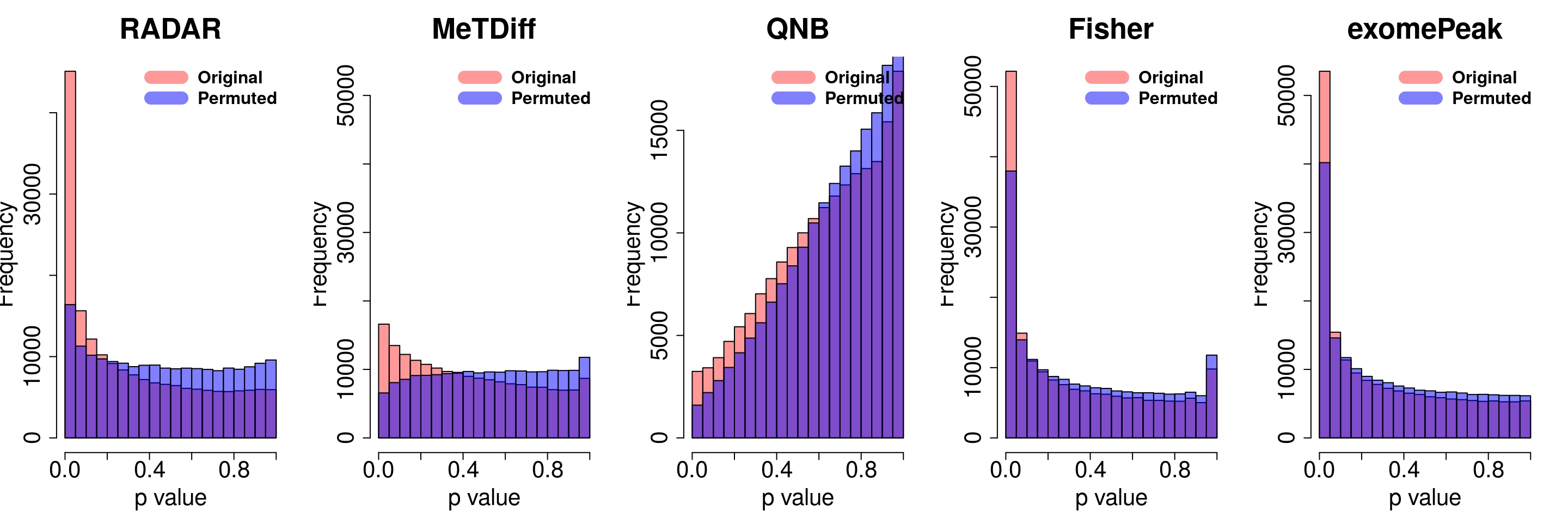
par(mfrow=c(1,5))
y.scale <- max(hist(exomePeak.res$pvalue,plot = F)$counts)
hist(RADAR@test.est[,"p_value"], col =rgb(1,0,0,0.4),main = "RADAR", xlab = "p value",font=2, cex.lab=2.5, cex.axis = 1.5 , font.lab=2 ,cex.main = 3, lwd = 3, ylim = c(0,y.scale) )
tmp <- hist(c(radar_permuted_P),plot = F)
tmp$counts <- tmp$counts/15
plot(tmp,col=rgb(0,0,1,0.5),add = T)
legend("topright", c("Original", "Permuted"), col=c(rgb(1,0,0,0.4), rgb(0,0,1,0.5)), lwd = 12,bty="n",text.font = 2, cex = 1.5)
tmp <- hist(c(metdiff_permuted_P),plot = F)
tmp$counts <- tmp$counts/15
hist(Metdiff.res$pvalue, col =rgb(1,0,0,0.4),main = "MeTDiff",xlab = "p value" ,font=2, cex.lab=2.5, cex.axis = 1.5 , font.lab=2 ,cex.main = 3, lwd = 3, ylim = c(0,y.scale) )
plot(tmp,col=rgb(0,0,1,0.5),add = T)
legend("topright", c("Original", "Permuted"), col=c(rgb(1,0,0,0.4), rgb(0,0,1,0.5)), lwd = 12,bty="n",text.font = 2, cex = 1.5)
hist(QNB.res$pvalue, col =rgb(1,0,0,0.4),main = "QNB",xlab = "p value",font=2, cex.lab=2.5, cex.axis = 1.5 , font.lab=2 ,cex.main = 3,lwd = 3,ylim = c(0,y.scale))
tmp <- hist(c(qnb_permuted_P),plot = F)
tmp$counts <- tmp$counts/15
plot(tmp,col=rgb(0,0,1,0.5),add = T)
legend("topright", c("Original", "Permuted"), col=c(rgb(1,0,0,0.4), rgb(0,0,1,0.5)), lwd = 12,bty="n",text.font = 2, cex = 1.5)
hist(fisher.res$pvalue, col =rgb(1,0,0,0.4),main = "Fisher",xlab = "p value",font=2, cex.lab=2.5, cex.axis = 1.5 , font.lab=2 ,cex.main = 3,lwd = 3,ylim = c(0,y.scale))
tmp <- hist(c(fisher_permuted_P),plot = F)
tmp$counts <- tmp$counts/15
plot(tmp,col=rgb(0,0,1,0.5),add = T)
legend("topright", c("Original", "Permuted"), col=c(rgb(1,0,0,0.4), rgb(0,0,1,0.5)), lwd = 12,bty="n",text.font = 2, cex = 1.5)
hist(exomePeak.res$pvalue, col =rgb(1,0,0,0.4),main = "exomePeak",xlab = "p value",font=2, cex.lab=2.5, cex.axis = 1.5 , font.lab=2 ,cex.main = 3,lwd = 3,ylim = c(0,y.scale))
tmp <- hist(c(exomePeak_permute_P),plot = F)
tmp$counts <- tmp$counts/15
plot(tmp,col=rgb(0,0,1,0.5),add = T)
legend("topright", c("Original", "Permuted"), col=c(rgb(1,0,0,0.4), rgb(0,0,1,0.5)), lwd = 12,bty="n",text.font = 2, cex = 1.5)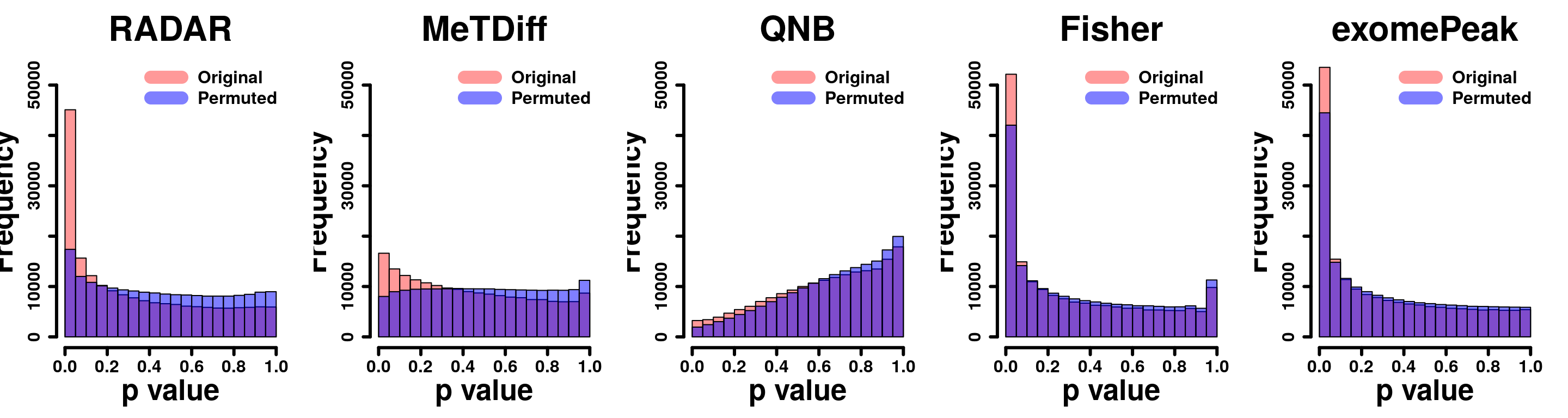
Permutation on position
radar_permuted_P_pos <-NULL
set.seed(51)
for(i in 1:15){
permute_X <- variable(RADAR)[,1]
newT2Did <- foreach(bat = unique(batch), .combine = c )%do%{
tmp_id <- which(batch %in% bat)
tmp_X <- variable(RADAR)[tmp_id,1]
return ( c(sample(x = tmp_id[which(tmp_X == "Ctl")], size = round(length(which(tmp_X == "Ctl"))/2) ),
sample(x = tmp_id[which(tmp_X == "T2D")], size = round(length(which(tmp_X == "T2D"))/2) )) )
}
permute_X[newT2Did] <- "T2D"
permute_X[setdiff(1:15,newT2Did)] <- "Ctl"
RADAR_permute <- RADAR_pos
variable(RADAR_permute) <- cbind( condition=permute_X, variable(RADAR_pos)[2:3] )
if ( all( permute_X == unlist(variable(RADAR)[,1]) ) | all( permute_X == c(rep("T2D",8),rep("Ctl",7))) ){
cat("Duplicated with original design...\n")
}else{
radar_permute <- diffIP_parallel( RADAR_permute , thread = 20 )@test.est[,"p_value"]
radar_permuted_P_pos <- cbind(radar_permuted_P_pos, radar_permute)
}
}par(mfrow=c(1,2))
y.scale <- max(hist( RADAR@test.est[,"p_value"] ,plot = F)$counts)
hist(RADAR@test.est[,"p_value"], col =rgb(1,0,0,0.4),main = "RADAR by geneSum", xlab = "p value",font=2, cex.lab=2.5, cex.axis = 1.5 , font.lab=2 ,cex.main = 3, lwd = 3, ylim = c(0,y.scale) )
tmp <- hist(c(radar_permuted_P),plot = F)
tmp$counts <- tmp$counts/15
plot(tmp,col=rgb(0,0,1,0.5),add = T)
legend("topright", c("Original", "Permuted"), col=c(rgb(1,0,0,0.4), rgb(0,0,1,0.5)), lwd = 12,bty="n",text.font = 2, cex = 1.5)
hist(RADAR_pos@test.est[,"p_value"], col =rgb(1,0,0,0.4),main = "RADAR by pos", xlab = "p value",font=2, cex.lab=2.5, cex.axis = 1.5 , font.lab=2 ,cex.main = 3, lwd = 3, ylim = c(0,y.scale) )
tmp <- hist(c(radar_permuted_P_pos),plot = F)
tmp$counts <- tmp$counts/15
plot(tmp,col=rgb(0,0,1,0.5),add = T)
legend("topright", c("Original", "Permuted"), col=c(rgb(1,0,0,0.4), rgb(0,0,1,0.5)), lwd = 12,bty="n",text.font = 2, cex = 1.5)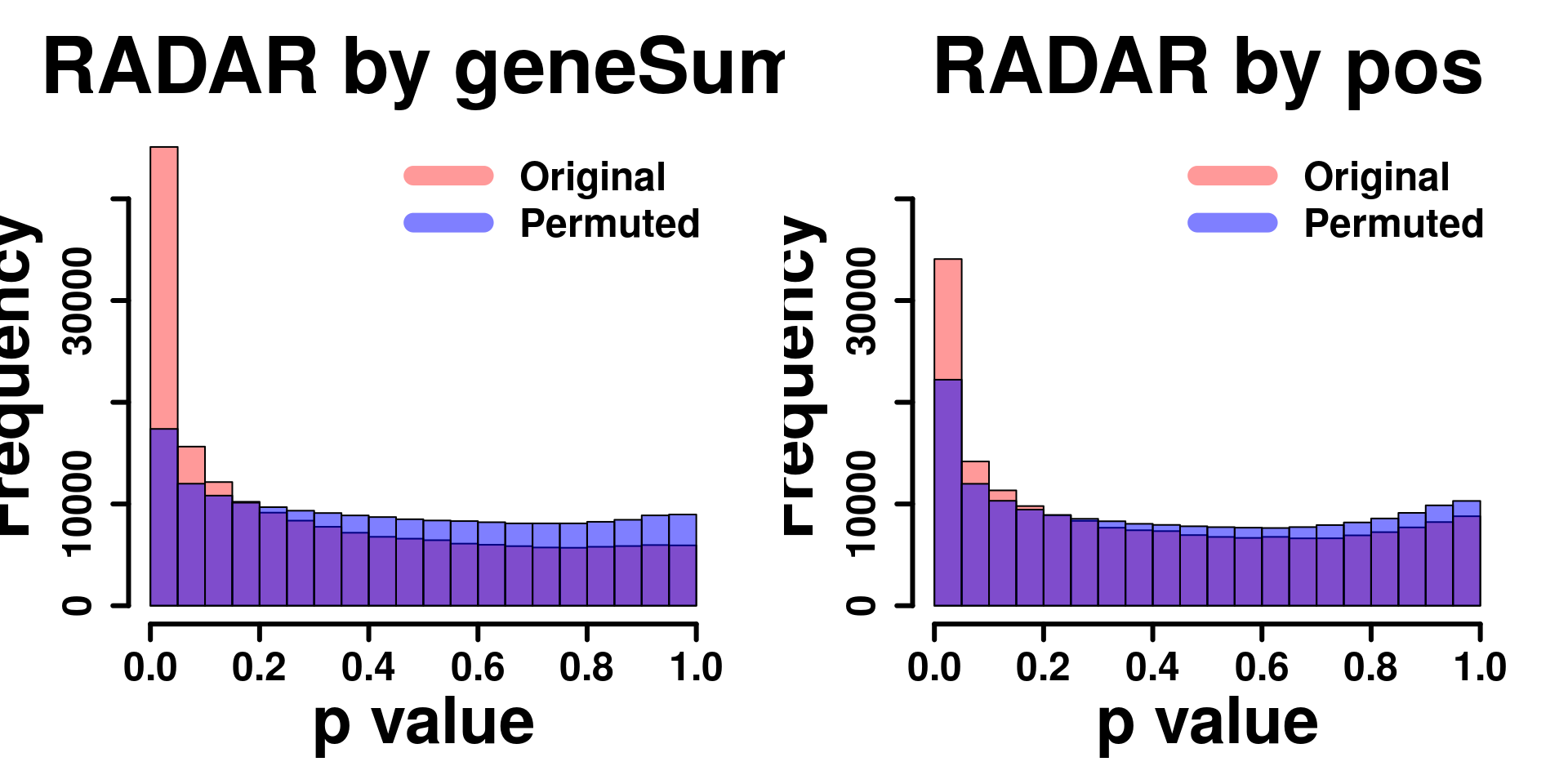
Plot Heat map of significant bins by RADAR
topBins <- RADAR@ip_adjExpr_filtered[which(RADAR@test.est[,"fdr"]<0.1),]
log_topBins <- log(topBins+1)
registerDoParallel(cores = 4)
cov.out.RADAR <- foreach(i = 1:nrow(log_topBins), .combine = rbind) %dopar% {
Y = log_topBins[i,]
tmp_data <- as.data.frame(cbind(Y,cov))
resi <- residuals( lm(Y~.,data=tmp_data ) )
resi
}
rm(list=ls(name=foreach:::.foreachGlobals), pos=foreach:::.foreachGlobals)
rownames(cov.out.RADAR) <- rownames(log_topBins)
colnames(cov.out.RADAR) <- colnames(log_topBins)
dist.pear <- function(x) as.dist(1-cor(t(x)))
hclust.ave <- function(x) hclust(x, method="average")
gplots::heatmap.2(cov.out.RADAR,scale="row",trace="none",labRow=NA,main = "Methylation level of significant bins\n(Covariates regressed out)",
distfun=dist.pear, hclustfun=hclust.ave,col=rev(RColorBrewer::brewer.pal(9,"RdBu")))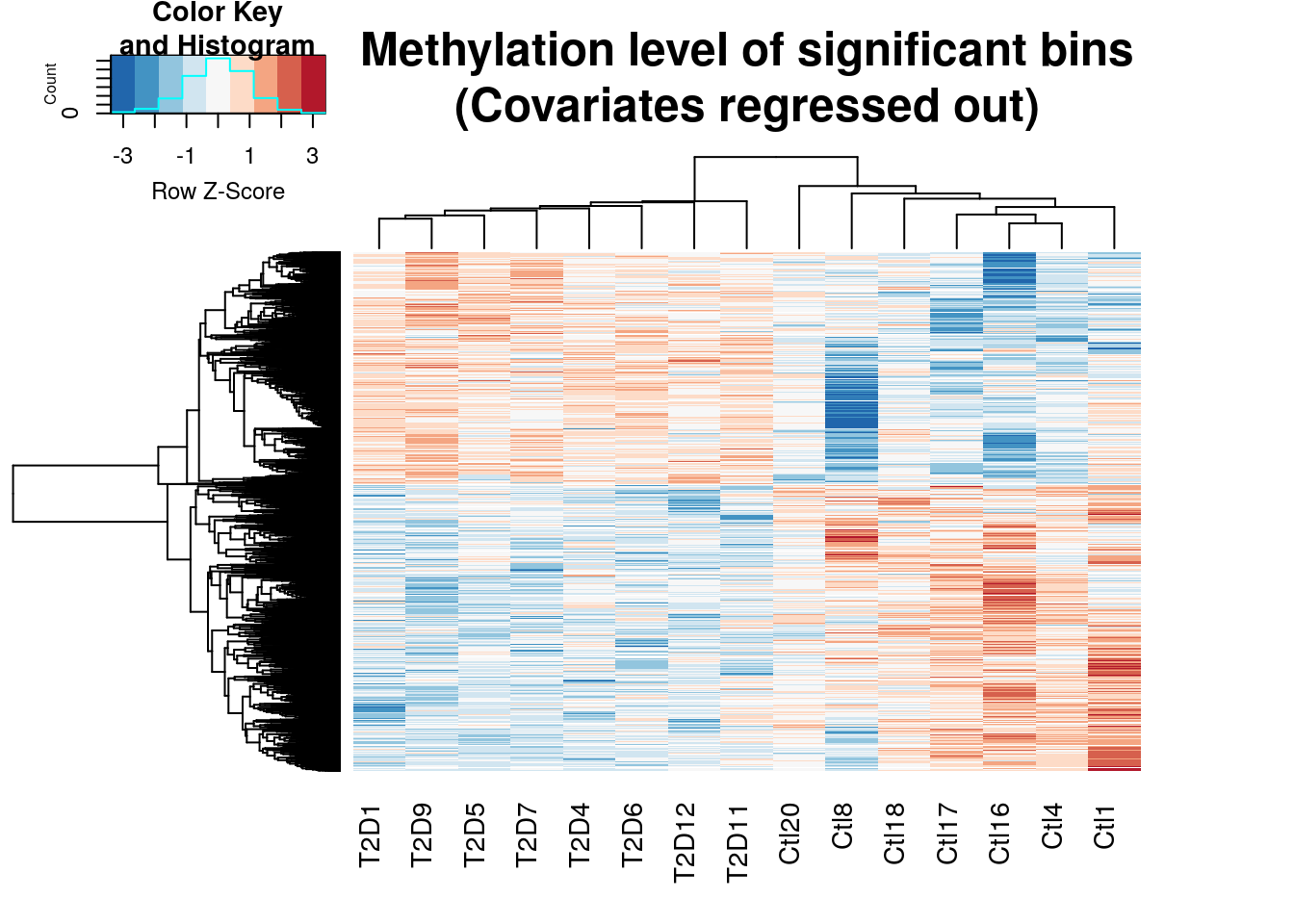
Report differential peaks
RADAR <- reportResult(RADAR, cutoff = 0.1,threads = 20)
RADAR_diff_peaks <- results(RADAR)
write.table(Diff_peaks_cov,"~/Tools/RADARmannual/data/T2D_RADAR_diffPeaks.xls",sep = "\t", col.names = T,row.names = F,quote = F)
reportPeaks <- function(radar, est , cutoff = 0.1, Beta_cutoff = 0.5, threads = 1){
stats <- est
rownames(stats) <- rownames(radar@test.est)
colnames(stats) <- gsub("padj","fdr",colnames(stats))
colnames(stats) <- gsub("log2.OR","beta",colnames(stats))
colnames(stats) <- gsub("logFC","beta",colnames(stats))
colnames(stats) <- gsub("log2.OR","beta",colnames(stats))
colnames(stats) <- gsub("pvalue","p_value",colnames(stats))
num_bins <- length( which(stats[,"fdr"] < cutoff & abs(stats[,"beta"] )> Beta_cutoff) )
if(num_bins < 1 ){
stop("There is no bin passing the threshold...\n No differential peaks can be reported at current cutoff...")
}else {
sig.bins <- rownames(stats)[which(stats[,"fdr"] < cutoff & abs(stats[,"beta"] )> Beta_cutoff)]
}
geneBins <- geneBins(radar)
ID <- (rownames(geneBins) %in% sig.bins)
num_lines <- length(ID)
# start ids of checkpoints
## find peak-starting checkpoints (either from nonpeak to peak, or peak in a different batch)
start_id <- which((ID[2:num_lines]-ID[1:num_lines-1]==1) |
((geneBins$gene[2:num_lines]!=geneBins$gene[1:num_lines-1]) & (ID[2:num_lines] == TRUE)) )
start_id <- start_id + 1 # add 1 since ID was counted from 2 to num_lines
if ( ID[1]==TRUE ) { start_id <- c(1,start_id) } # if the first checkpoint bin is peak
# end ids of checkpoints
## find peak-ending checkpoints (either from peak to nonpeak, or peak in a different batch)
end_id <- which((ID[1:num_lines-1]-ID[2:num_lines]==1) |
((geneBins$gene[1:num_lines-1]!=geneBins$gene[2:num_lines]) & (ID[1:num_lines-1] == TRUE)) )
if (ID[num_lines]==TRUE) {end_id <- c(end_id,num_lines)} # if the last checkpoint bin is peak
peak_id_pairs <- cbind(start_id, end_id)
num_peaks <- nrow(peak_id_pairs)
geneGRList <- radar@geneModel
peakGenes <- as.character(geneBins[peak_id_pairs[,1],"gene"])
cat(paste0("reporting ",num_peaks," of peaks\n"))
if (num_peaks == 0){return(data.frame())
}else{
start_time <- Sys.time()
registerDoParallel(cores = threads)
cat(paste("Hyper-thread registered:",getDoParRegistered(),"\n"))
cat(paste("Using",getDoParWorkers(),"thread(s) to report merged report...\n"))
merged.report<- foreach( p = 1:num_peaks, .combine = rbind)%dopar%{
peak_row_id <- peak_id_pairs[p,]
geneExons <- reduce ( geneGRList[peakGenes[p]][[1]] )
peak <- RADAR:::.getPeakBins(geneGRList,peakGenes[p],c(geneBins$bin[peak_row_id[1]],geneBins$bin[peak_row_id[2]]),radar@binSize )
peakE <- RADAR:::.peakExons(peak,as.data.frame(geneExons))
data.frame(chr=peak$chr,
start = peak$start,
end = peak$end,
name = peakGenes[p],
score = 0,
strand = as.character(strand(geneExons))[1],
thickStart = peak$start,
thickEnd = peak$end,
itemRgb=0,
blockCount = nrow(peakE),
blockSizes = paste(peakE$width,collapse=","),
blockStarts = paste(peakE$start - replicate(nrow(peakE),peakE$start[1]),collapse=","),
logFC = stats[rownames(geneBins[peak_row_id[1]:peak_row_id[2],]), "beta"][which.max(abs( stats[rownames(geneBins[peak_row_id[1]:peak_row_id[2],]), "beta"] ))],
p_value = RADAR:::.fishersMethod( stats[rownames(geneBins[peak_row_id[1]:peak_row_id[2],]), "p_value"] )
)
}
rm(list=ls(name=foreach:::.foreachGlobals), pos=foreach:::.foreachGlobals)
end_time <- Sys.time()
cat(paste("Time used to report peaks:",difftime(end_time, start_time, units = "mins"),"mins... \n"))
}
return(merged.report)
}
exomePeak_diff <- reportPeaks(RADAR, est = exomePeak.res, threads = 20)
QNB_diff <- reportPeaks(RADAR, est = QNB.res, threads = 20)
Metdiff_diff <- reportPeaks(RADAR, est = Metdiff.res, threads = 20)
fisher_diff <- reportPeaks(RADAR, est = fisher.res, threads = 20)library(MeRIPtools)
plotMetaGeneMulti(list("RADAR" = RADAR_diff_peaks,"exomePeak"=exomePeak_diff, "Fisher" = fisher_diff ),"~/Database/genome/hg38/hg38_UCSC.gtf")## [1] "Converting BED12 to GRangesList"
## [1] "It may take a few minutes"
## [1] "Converting BED12 to GRangesList"
## [1] "It may take a few minutes"
## [1] "Converting BED12 to GRangesList"
## [1] "It may take a few minutes"
## [1] "total 58259 transcripts extracted ..."
## [1] "total 42543 transcripts left after ambiguity filter ..."
## [1] "total 21293 mRNAs left after component length filter ..."
## [1] "total 7986 ncRNAs left after ncRNA length filter ..."
## [1] "Building Guitar Coordinates. It may take a few minutes ..."
## [1] "Guitar Coordinates Built ..."
## [1] "Using provided Guitar Coordinates"
## [1] "resolving ambiguious features ..."
## [1] "no figure saved ..."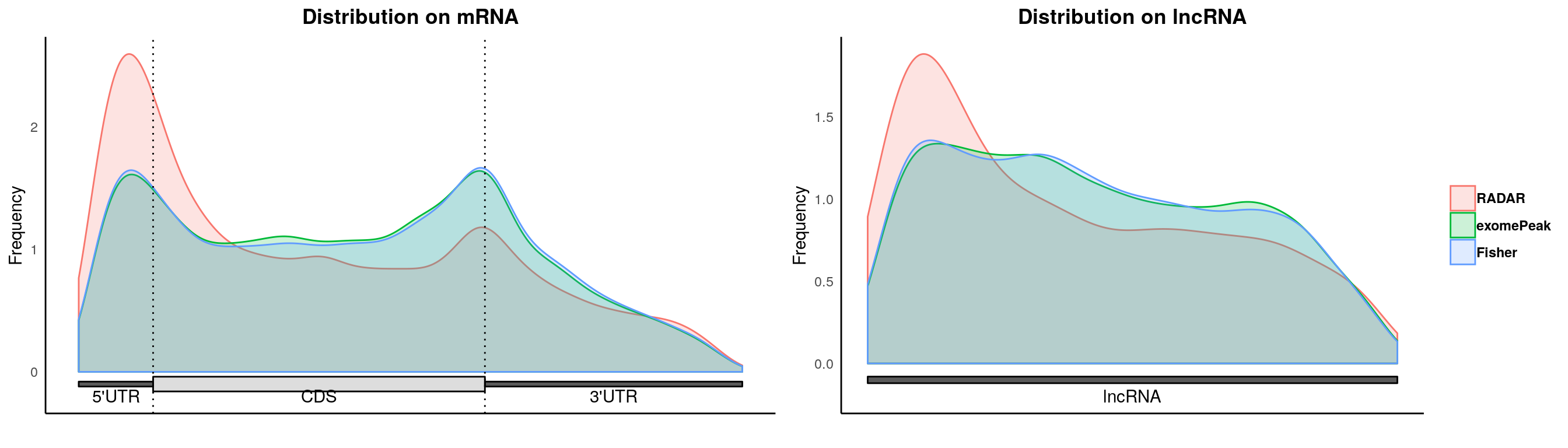
## NOTE this function is a wrapper for R package "Guitar".
## If you use the metaGene plot in publication, please cite the original reference:
## Cui et al 2016 BioMed Research Internationaldetach("package:MeRIPtools", unload = TRUE)extendPeak <- function(peak, extension = 50){
peak_extend <- peak
peak_extend$start <- peak$start-extension
peak_extend$end <- peak$end+extension
peak_extend$blockSizes <- unlist( lapply( strsplit(as.character(peak$blockSizes),split = ",") , function(x){
tmp = as.numeric(x)
tmp[1] = tmp[1]+extension
tmp[length(tmp)] = tmp[length(tmp)]+extension
paste(as.character(tmp),collapse = ",")
} ) )
return(peak_extend)
}
RADAR_diff_extend <- extendPeak(RADAR_diff_peaks)
exomePeak_diff_extend <- extendPeak(exomePeak_diff)
Metdiff_diff_extend <- extendPeak(Metdiff_diff)
QNB_diff_extend <- extendPeak(QNB_diff)
fisher_diff_extend <- extendPeak(fisher_diff)## analysis for RADAR detected signifiant bins
write.table(RADAR_diff_extend[,1:12], file = paste0("~/Tools/RADARmannual/data/T2D_Diff_peaks_ageCov.bed"),sep = "\t",row.names = F,col.names = F,quote = F)
system2(command = "bedtools", args = paste0("getfasta -fi ~/Database/genome/hg38/hg38_UCSC.fa -bed ~/Tools/RADARmannual/data/T2D_Diff_peaks_ageCov.bed -s -split > ~/Tools/RADARmannual/data/T2D_Diff_peaks_ageCov.fa ") )
system2(command = "findMotifs.pl", args = paste0("~/Tools/RADARmannual/data/T2D_Diff_peaks_ageCov.fa fasta ~/Tools/RADARmannual/data/T2D_Diff_peaks_ageCov_Homer2 -fasta ~/Database/transcriptome/backgroud_peaks/hg38_90bpRandomPeak.fa -len 5,6 -rna -p 20 -S 5 -noknown"),wait = F )
## analysis for exomePeak detected significant bins
write.table(exomePeak_diff_extend[,1:12], file = paste0("~/Tools/RADARmannual/data/T2Dr_Diff_exomePeak.bed"),sep = "\t",row.names = F,col.names = F,quote = F)
system2(command = "bedtools", args = paste0("getfasta -fi ~/Database/genome/hg38/hg38_UCSC.fa -bed ~/Tools/RADARmannual/data/T2Dr_Diff_exomePeak.bed -s -split > ~/Tools/RADARmannual/data/T2Dr_Diff_exomePeak.fa ") )
system2(command = "findMotifs.pl", args = paste0("~/Tools/RADARmannual/data/T2Dr_Diff_exomePeak.fa fasta ~/Tools/RADARmannual/data/T2Dr_Diff_exomePeak_Homer2 -fasta ~/Database/transcriptome/backgroud_peaks/hg38_90bpRandomPeak.fa -len 5,6 -rna -p 20 -S 5 -noknown"),wait = F )
## analysis for MetDiff detected significant bins
write.table(Metdiff_diff_extend[,1:12], file = paste0("~/Tools/RADARmannual/data/T2Dr_Diff_MetDiff.bed"),sep = "\t",row.names = F,col.names = F,quote = F)
system2(command = "bedtools", args = paste0("getfasta -fi ~/Database/genome/hg38/hg38_UCSC.fa -bed ~/Tools/RADARmannual/data/T2Dr_Diff_MetDiff.bed -s -split > ~/Tools/RADARmannual/data/T2Dr_Diff_MetDiff.fa ") )
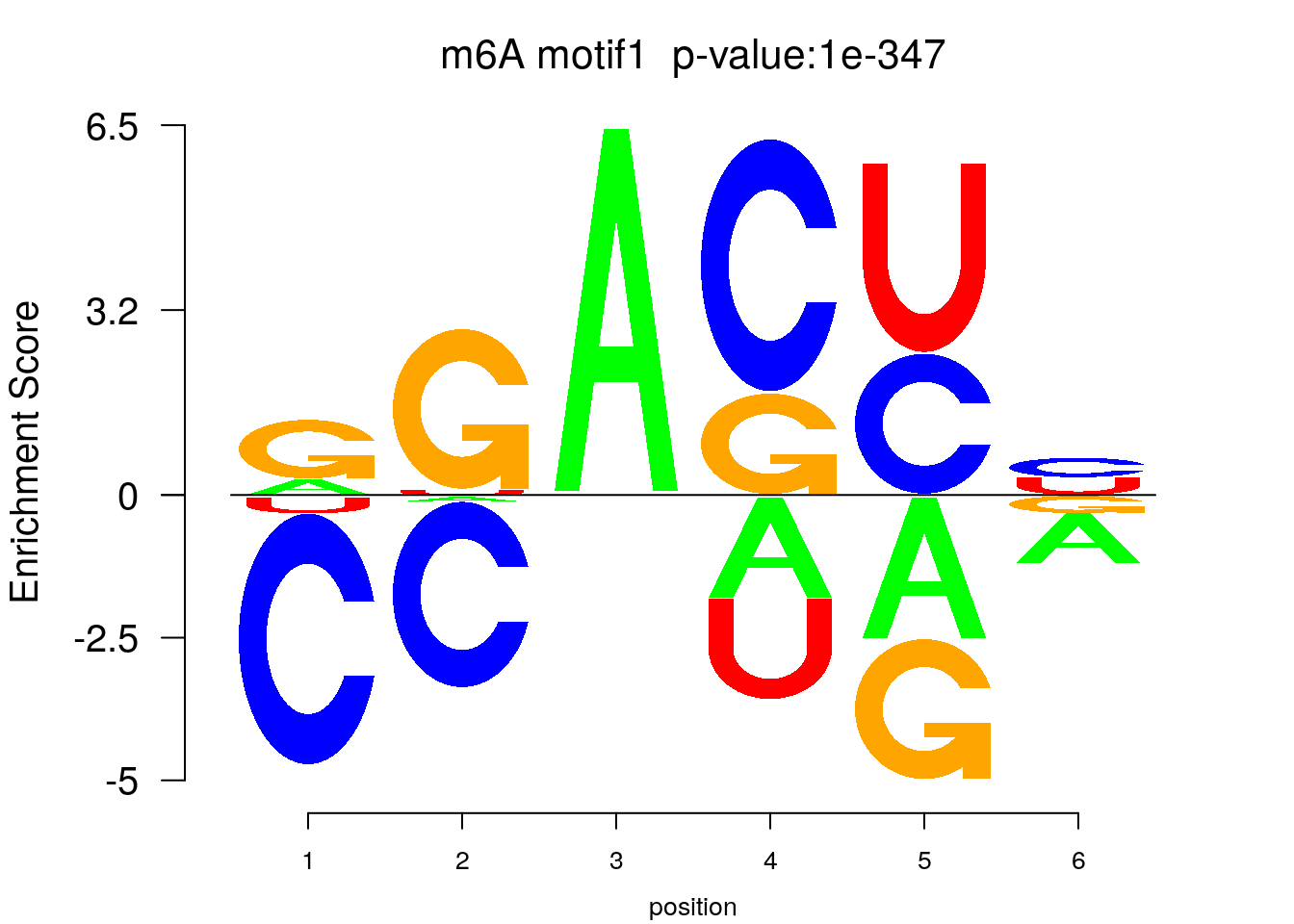
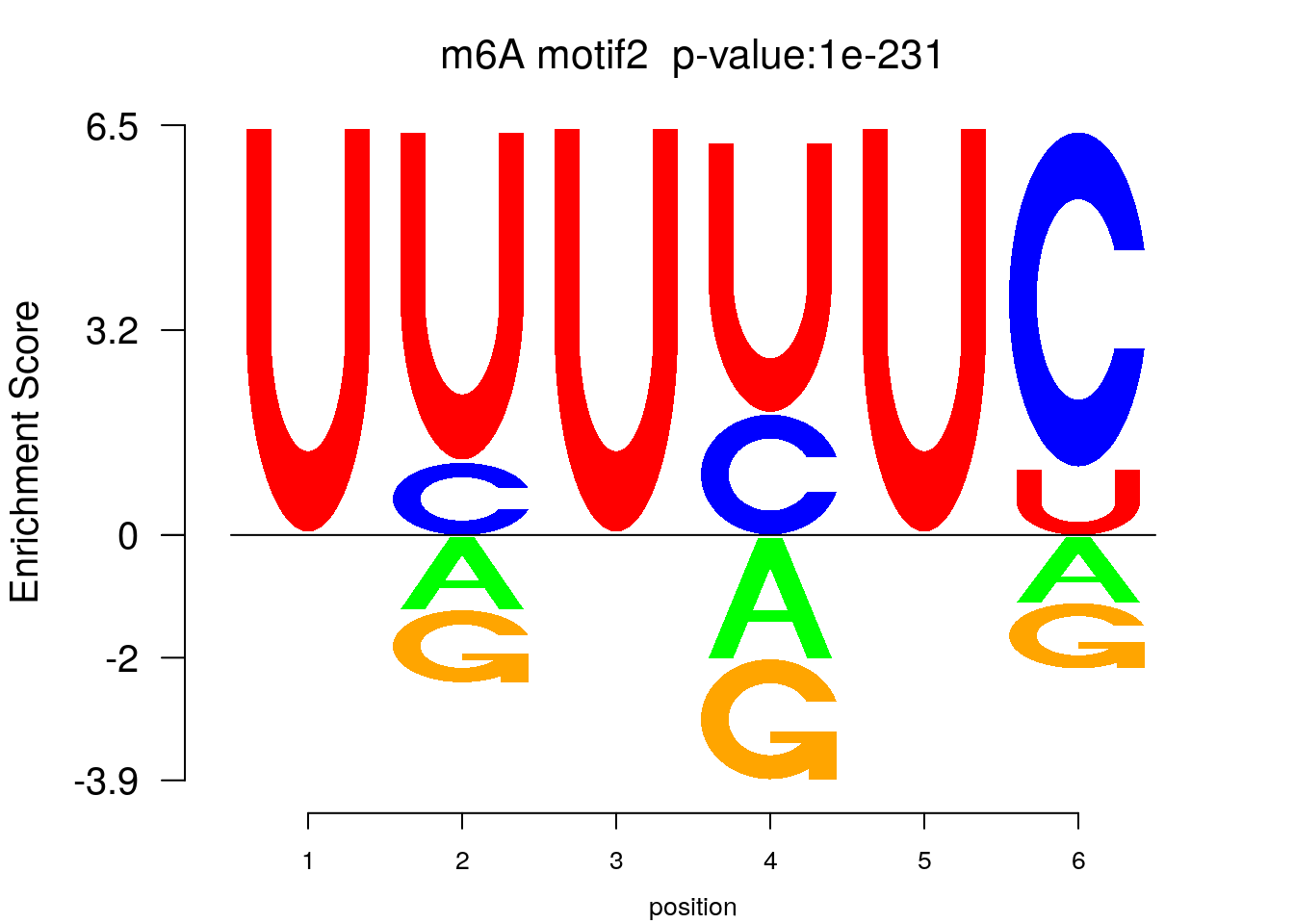
system2(command = "findMotifs.pl", args = paste0("~/Tools/RADARmannual/data/T2Dr_Diff_MetDiff.fa fasta ~/Tools/RADARmannual/data/T2Dr_Diff_MetDiff_Homer2 -fasta ~/Database/transcriptome/backgroud_peaks/hg38_90bpRandomPeak.fa -len 5,6 -rna -p 20 -S 5 -noknown"),wait = F )library(Logolas)
color_motif <- c( "orange", "blue", "red","green" )
for(i in 1:3){
pwm.m <- t( read.table(paste0("~/Tools/RADARmannual/data/T2D_Diff_peaks_ageCov_Homer2/homerResults/motif",i,".motif"), header = F, comment.char = ">",col.names = c("A","C","G","U")) )
motif_p <- sapply(1:3, function(x){
strsplit( as.character( read.table(paste0("~/Tools/RADARmannual/data/T2D_Diff_peaks_ageCov_Homer2/homerResults/motif",x,".motif"),comment.char = "", nrows = 1)[,12] ), split= ":")[[1]][4]
})
colnames(pwm.m) <- 1:ncol(pwm.m)
try(logomaker(pwm.m ,type = "EDLogo",colors = color_motif,color_type = "per_row" ,logo_control = list(pop_name = paste0("m6A motif",i," p-value:",motif_p[i])))
)
}

## Error in grid::unit(letters$x, "native") :
## 'x' and 'units' must have length > 0RADAR_diff_extend.gr <- makeGRangesFromDataFrame(RADAR_diff_extend)
QNB_diff_extend.gr <- makeGRangesFromDataFrame(QNB_diff_extend)
exomePeak_diff_extend.gr <- makeGRangesFromDataFrame(exomePeak_diff_extend)
Metdiff_diff_extend.gr <- makeGRangesFromDataFrame(Metdiff_diff_extend)
fisher_diff_extend.gr <- makeGRangesFromDataFrame(fisher_diff_extend)
R_all_overlap <- findOverlaps( RADAR_diff_extend.gr , reduce(c(QNB_diff_extend.gr, Metdiff_diff_extend.gr,exomePeak_diff_extend.gr,fisher_diff_extend.gr)) )
RADAR_specific <- RADAR_diff_extend[ ! 1:length(RADAR_diff_extend.gr)%in% queryHits(R_all_overlap),]
E_F_R_overlap <- findOverlaps( exomePeak_diff_extend.gr[unique( queryHits( findOverlaps(exomePeak_diff_extend.gr,fisher_diff_extend.gr) ) )], RADAR_diff_extend.gr)
bogus_specific <- exomePeak_diff_extend[ ! 1:length(exomePeak_diff_extend.gr) %in% queryHits(E_F_R_overlap) , ]RADAR specific DM site RADAR pvalue 2.84e-7, beta = -0.66
plotGeneCov(RADAR,"RNF213" ,ZoomIn = c(80395388,80395837),adjustExprLevel = TRUE)+scale_x_continuous(expand = c(0.02,0))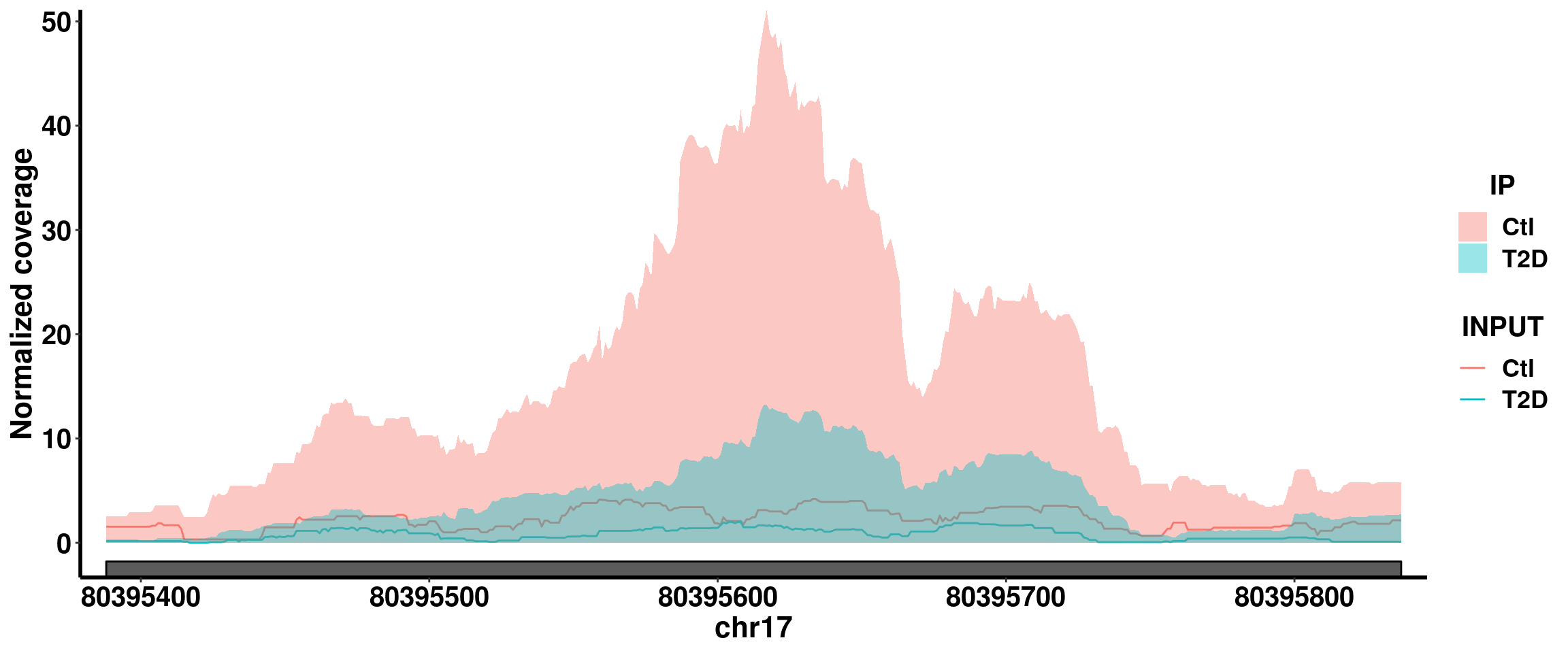
plotGeneCov(RADAR,"RNF213" ,ZoomIn = c(80395388,80395837),adjustExprLevel = TRUE, split = TRUE)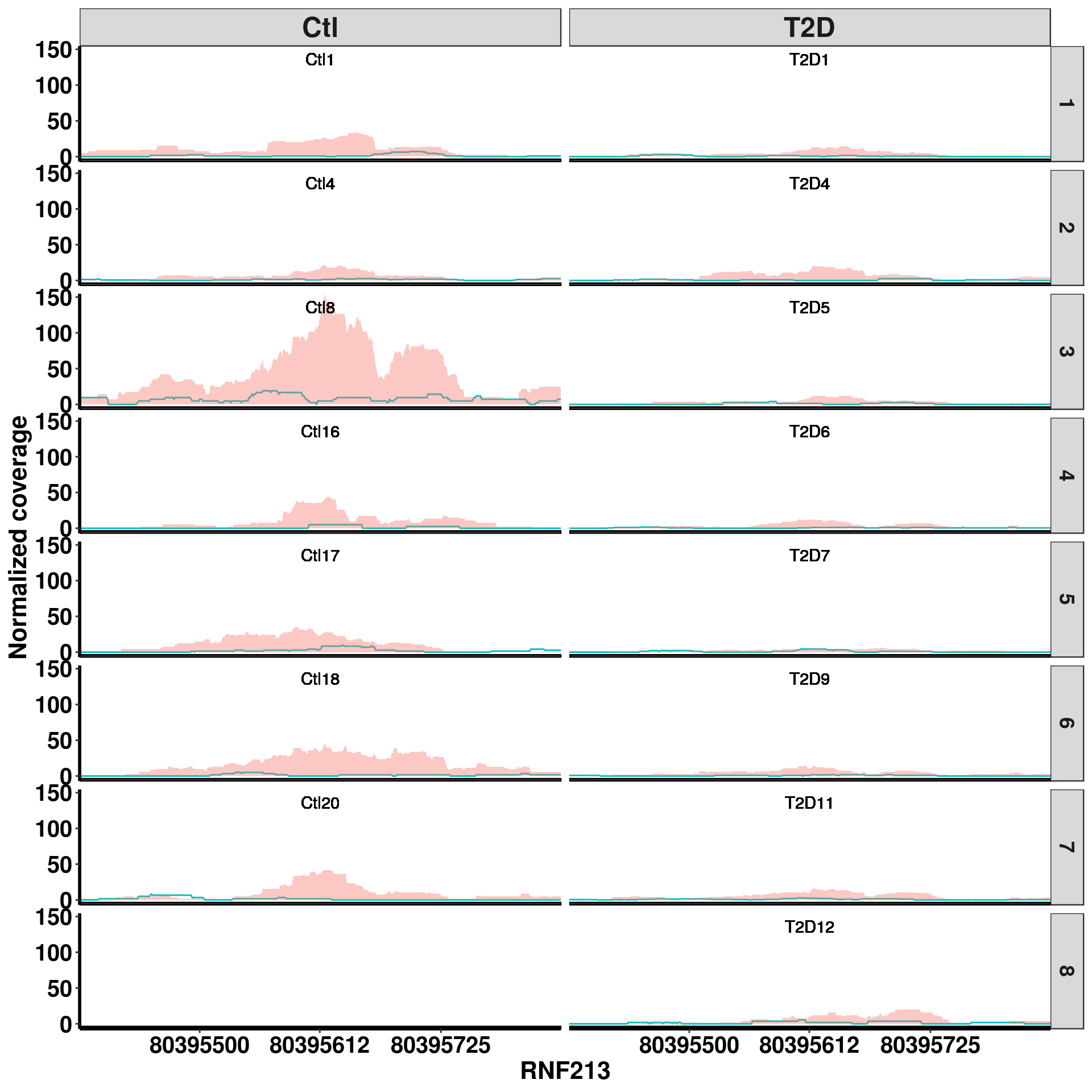
Bogus DM sites exomePeak pvalue 1.07e-9, beta = 0.71
plotGeneCov(RADAR,"POLR2A" ,ZoomIn = c(7511258,7511493),adjustExprLevel = T, split = FALSE)+scale_x_continuous(expand = c(0.02,0))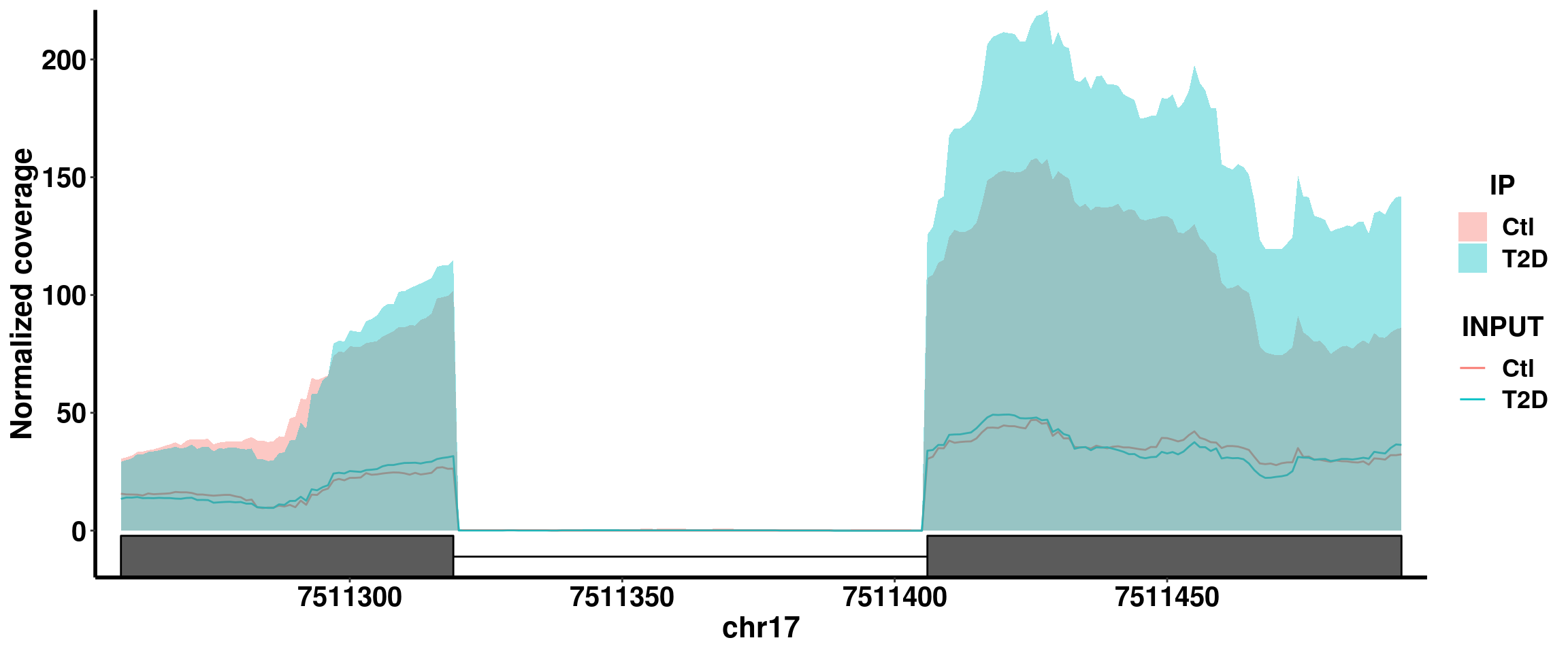
plotGeneCov(RADAR,"POLR2A" ,ZoomIn = c(7511258,7511493),adjustExprLevel = T, split = TRUE)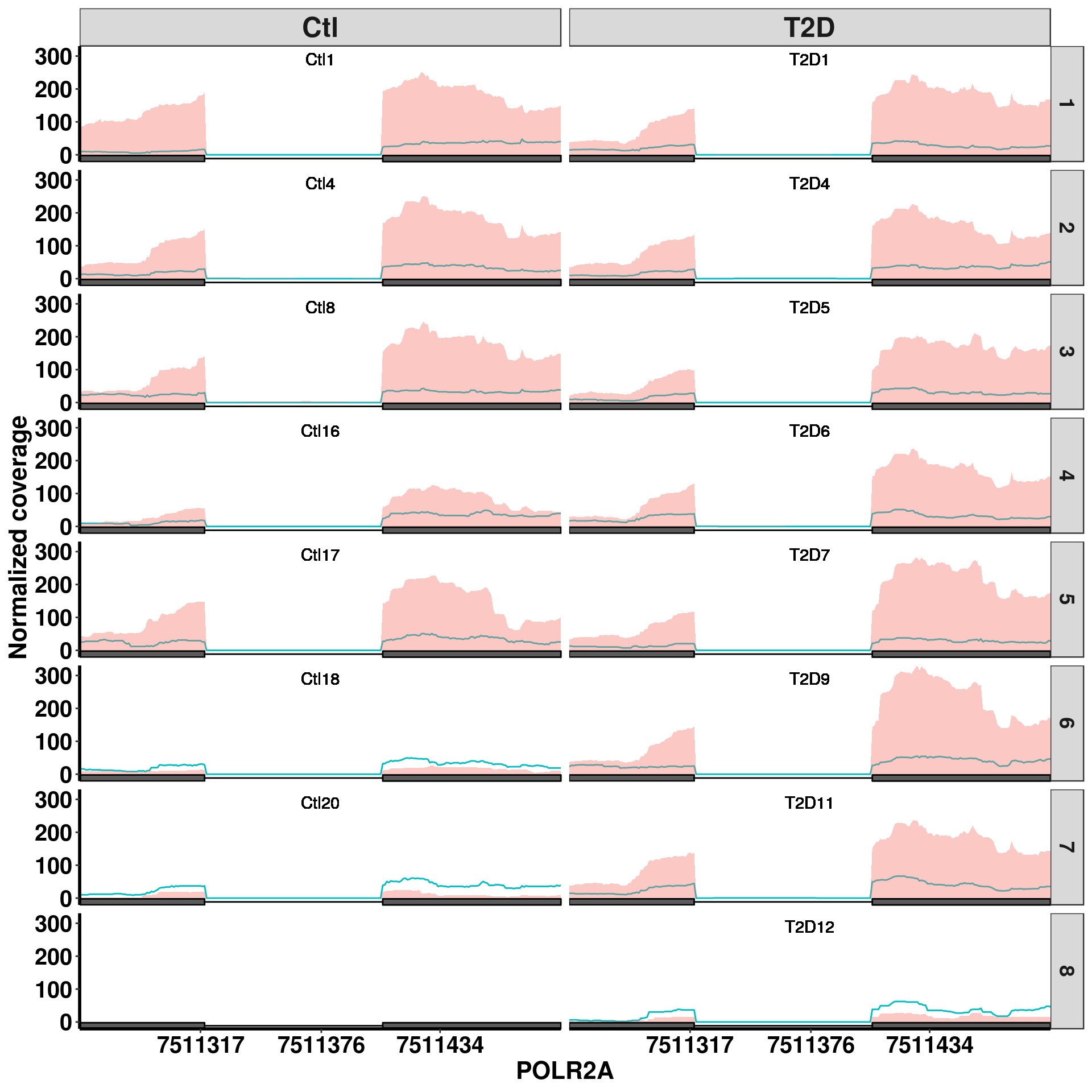
Bogus DM sites exomePeak report this site at P-value 2.367657e-06 for up regulation in T2D, beta = 0.654. Splited coverage plot indicates it is mainly driven by T2D4 sample, but not others.
plotGeneCov(RADAR,"ABAT" ,ZoomIn = c(8781835,8781984),adjustExprLevel = TRUE, split = FALSE)+scale_x_continuous(expand = c(0.02,0))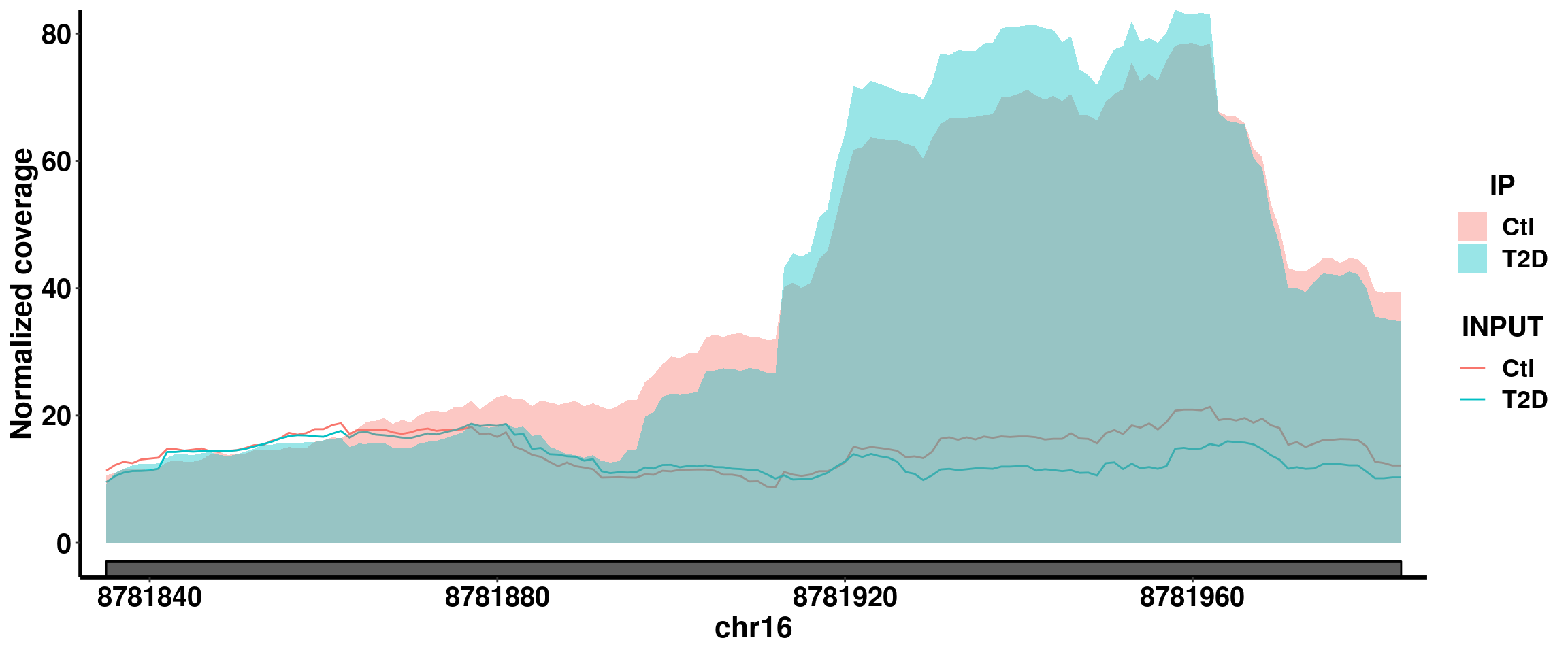
plotGeneCov(RADAR,"ABAT" ,ZoomIn = c(8781835,8781984),adjustExprLevel = TRUE, split = TRUE)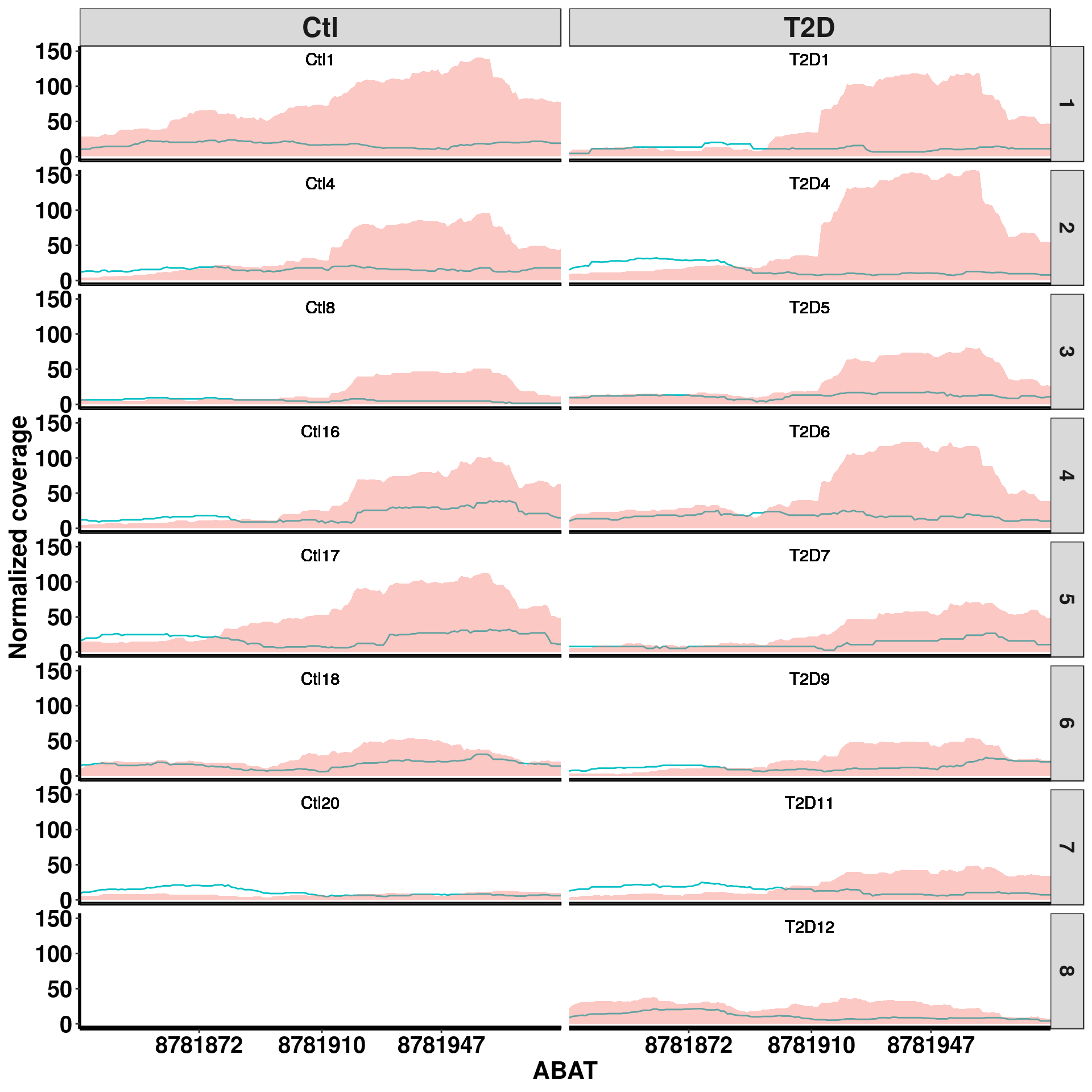
Session information
sessionInfo()## R version 3.5.3 (2019-03-11)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 17.10
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/libopenblasp-r0.2.20.so
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] grid stats4 parallel stats graphics grDevices utils
## [8] datasets methods base
##
## other attached packages:
## [1] Logolas_1.6.0 reshape2_1.4.3
## [3] GenomicAlignments_1.18.1 SummarizedExperiment_1.12.0
## [5] DelayedArray_0.8.0 BiocParallel_1.16.6
## [7] matrixStats_0.54.0 rtracklayer_1.42.2
## [9] RADAR_0.3.0 qvalue_2.14.1
## [11] RcppArmadillo_0.9.400.2.0 Rcpp_1.0.1
## [13] RColorBrewer_1.1-2 gplots_3.0.1.1
## [15] doParallel_1.0.14 iterators_1.0.10
## [17] foreach_1.4.4 ggplot2_3.1.1
## [19] Rsamtools_1.34.1 Biostrings_2.50.2
## [21] XVector_0.22.0 GenomicFeatures_1.34.8
## [23] AnnotationDbi_1.44.0 Biobase_2.42.0
## [25] GenomicRanges_1.34.0 GenomeInfoDb_1.18.2
## [27] IRanges_2.16.0 S4Vectors_0.20.1
## [29] BiocGenerics_0.28.0
##
## loaded via a namespace (and not attached):
## [1] backports_1.1.4 Hmisc_4.2-0 fastmatch_1.1-0
## [4] plyr_1.8.4 igraph_1.2.4.1 lazyeval_0.2.2
## [7] splines_3.5.3 gamlss_5.1-3 gridBase_0.4-7
## [10] urltools_1.7.3 digest_0.6.18 htmltools_0.3.6
## [13] GOSemSim_2.8.0 viridis_0.5.1 GO.db_3.7.0
## [16] SQUAREM_2017.10-1 gdata_2.18.0 magrittr_1.5
## [19] checkmate_1.9.1 memoise_1.1.0 BSgenome_1.50.0
## [22] cluster_2.0.7-1 annotate_1.60.1 enrichplot_1.2.0
## [25] prettyunits_1.0.2 colorspace_1.4-1 blob_1.1.1
## [28] ggrepel_0.8.0 gamlss.data_5.1-3 exomePeak_2.16.0
## [31] xfun_0.6 dplyr_0.8.0.1 crayon_1.3.4
## [34] RCurl_1.95-4.12 jsonlite_1.6 genefilter_1.64.0
## [37] ape_5.3 survival_2.44-1.1 glue_1.3.1
## [40] polyclip_1.10-0 gtable_0.3.0 zlibbioc_1.28.0
## [43] UpSetR_1.3.3 scales_1.0.0 DOSE_3.8.2
## [46] DBI_1.0.0 viridisLite_0.3.0 xtable_1.8-4
## [49] progress_1.2.0 htmlTable_1.13.1 gridGraphics_0.3-0
## [52] foreign_0.8-71 bit_1.1-14 europepmc_0.3
## [55] Formula_1.2-3 htmlwidgets_1.3 httr_1.4.0
## [58] fgsea_1.8.0 acepack_1.4.1 pkgconfig_2.0.2
## [61] XML_3.98-1.19 farver_1.1.0 nnet_7.3-12
## [64] locfit_1.5-9.1 ggplotify_0.0.3 tidyselect_0.2.5
## [67] labeling_0.3 rlang_0.4.0 munsell_0.5.0
## [70] tools_3.5.3 generics_0.0.2 RSQLite_2.1.1
## [73] broom_0.5.2 ggridges_0.5.1 evaluate_0.13
## [76] stringr_1.4.0 yaml_2.2.0 knitr_1.22
## [79] bit64_0.9-7 fs_1.3.0 caTools_1.17.1.2
## [82] purrr_0.3.2 ggraph_1.0.2 nlme_3.1-137
## [85] QNB_1.1.11 DO.db_2.9 xml2_1.2.0
## [88] biomaRt_2.38.0 compiler_3.5.3 rstudioapi_0.10
## [91] Guitar_1.20.1 tibble_2.1.1 tweenr_1.0.1
## [94] geneplotter_1.60.0 stringi_1.4.3 lattice_0.20-38
## [97] Matrix_1.2-17 permute_0.9-5 vegan_2.5-4
## [100] pillar_1.3.1 triebeard_0.3.0 data.table_1.12.2
## [103] cowplot_0.9.4 bitops_1.0-6 R6_2.4.0
## [106] latticeExtra_0.6-28 vcfR_1.8.0 KernSmooth_2.23-15
## [109] gridExtra_2.3 codetools_0.2-16 MASS_7.3-51.4
## [112] gtools_3.8.1 assertthat_0.2.1 DESeq2_1.22.2
## [115] pinfsc50_1.1.0 withr_2.1.2 GenomeInfoDbData_1.2.0
## [118] mgcv_1.8-28 hms_0.4.2 clusterProfiler_3.10.1
## [121] rpart_4.1-13 tidyr_0.8.3 rmarkdown_1.12
## [124] rvcheck_0.1.3 ggforce_0.2.2 gamlss.dist_5.1-3
## [127] base64enc_0.1-3This R Markdown site was created with workflowr